
Abstract
Calgranulins are small calcium-binding proteins with several immunological functions involved in inflammatory processes. Calgranulin A is reported to be mainly associated with acute inflammation while calgranulin B seems to play a role in chronic inflammatory disorders. In this study we used a proteomic approach to analyse calgranulin B expression in bronchoalveolar lavage (BAL) from a group of patients with different interstitial lung diseases. Two dimensional electrophoresis analysis of BAL was performed in 11 idiopathic pulmonary fibrosis patients, nine sarcoidosis patients, 11 with systemic sclerosis patients and five healthy controls. Significantly higher (p < 0.001) calgranulin B percentage volumes were observed in BAL from IPF patients than controls and other ILD patients. This result sustains the hypothesis that calgranulin B could be involved in chronic lung diseases, probably through increased expression and enhanced activation of alveolar polymorphonuclear cells related to idiopathic pulmonary fibrosis. Quantitative analysis by an easier method applied to a larger population will be necessary to determine whether calgranulin B could be a good marker of disease severity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Calgranulins are members of the S100 family of small calcium-binding proteins [1] and are linked to innate immune functions by expression in cells such as monocytes, neutrophils and early differentiation stages of macrophages [2, 3]. They have a role in migration and cytoskeletal metabolism. After release into the extracellular space they activate immune cells and vascular endothelium causing cell damage. Their role in chronic lung inflammation is subject of much research [4]. Calgranulin expression has been analysed in different autoimmune diseases including lung disorders [1, 4]. These S100 proteins have been associated with the pathogenesis of cystic fibrosis, a disease in which neutrophils are a major mediator of lung damage. They have also been measured in sputum from patients with COPD and BAL from ARDS patients.
We recently analysed the protein composition of bronchoalveolar lavage (BAL) from idiopathic pulmonary fibrosis (IPF) and sarcoidosis (S) patients by a proteomic approach [5]. In 2002, our group reported the BAL protein pattern of six patients with sarcoidosis and six patients with IPF studied by two dimensional electrophoresis (2-DE) [5]. Certain proteins identified by gel matching and MALDI-TOF mass fingerprinting were expressed differently in the two diseases. In particular, comparison of bronchoalveolar lavage protein maps of the two groups of patients showed 32 spots with statistically significant disease-related variations in relative abundance. Among these, S100A8 (calgranulin A) and S100A9 (Calgranulin B) were first identified in BAL of these patients and their percentage volumes were significantly increased in IPF with respect to sarcoidosis patients [5]. In 2005 we analysed BAL protein composition in patients with different ILD; more recently BAL protein composition from ILD patients was compared with a group of controls [6].
The aim of the present study was to analyse calgranulin B expression in a group of patients with idiopathic pulmonary fibrosis in order to verify our previous findings and to compare calgranulin B percentage volumes in patients with IPF, sarcoidosis and pulmonary fibrosis associated with systemic sclerosis (Ssc) and controls.
Materials and Methods
The study population consisted of 11 IPF patients, nine sarcoidosis patients, 11 patients with pulmonary fibrosis associated with systemic sclerosis and five healthy controls. Sarcoidosis, pulmonary fibrosis associated with systemic sclerosis and IPF were diagnosed according to the International Consensus criteria [7, 8]. All subjects underwent bronchoscopy for clinical purposes. The patients with interstitial lung diseases included in the study had never been treated with steroids or other immunosuppressants at the time of bronchoalveolar lavage. They were regularly followed at the Sarcoidosis and Fibrosis Regional Referral Centre in Siena from onset for at least 12 months. All patients enrolled in the study underwent medical history, physical examination, chest X-ray, chest HRCT, lung function tests and DLCO, 6 min walking test, blood gas analysis and bronchoscopy with BAL. Personal, clinical and immunological data of patients and controls are reported in Table 1. No patient had acute onset, Löfgren disease or acute exacerbation of IPF/Ssc at the time of bronchoscopy. The group of controls consisted of five healthy subjects with no history of asthma, allergy or respiratory infections and normal lung function parameters. Patients and controls gave their written informed consent to the study.
Bronchoscopy and bronchoalveolar lavage were performed as previously described [5, 6]. In brief BAL was carried out by the standard method, instilling 5 × 60 ml aliquots of normal saline by fibre bronchoscope (Olympus IT-10; Olympus Italia, Milan, Italy). The first sample was kept separate from the others and was not used for immunological tests. Cells were separate by centrifuge from the fluid component of BAL that was frozen in aliquots for the study of protein composition. Cell differential count was performed. The phenotype of BAL lymphocytes was analysed by flow cytometry using monoclonal antibodies.
BAL samples were dialysed against water, lyophilised and dissolved in lysis buffer so that 100 μl of sample contained 45 μg of proteins. 2-DE was performed as previously described [9, 10].
Calgranulin B was analysed by two-dimensional electrophoresis, identified by gel matching and MALDI-TOF mass fingerprinting, as previously reported [10]. Electrophoretogram images were obtained with a computing densitometer and processed by the Melanie III computer system. For quantitative analysis we calculated volumes with respect to the sum of the volumes of all spots in each gel in order to correct for variability related to silver staining.
Statistical analysis of data was performed using StatSoft 2001 software for Windows.
Results and Discussion
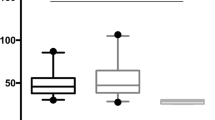
Personal data, spirometric values and BAL features from patients and controls are reported in Table 1. Calgranulin B percentage volumes in BAL of patients and controls are described in Fig. 1. Significantly higher calgranulin B percentage volumes were found in BAL of IPF patients than controls (p < 0.01), sarcoidosis patients (p < 0.01) and Ssc patients (p < 0.01) (Fig. 1). In healthy controls calgranulin B percentage volumes were similar to those of sarcoidosis and Ssc patients while a substantial increase was found in IPF patients. No correlations were found between calgranulin B percentage volumes and clinical parameters, including chest radiological stages of sarcoidosis and lung function results taken as indicators of disease severity. This may be due to the limited number of patients analysed and to the use of a semiquantitative test (2-DE rather an ELISA).

Percentage volumes of calgranulin B in BAL of sarcoidosis, pulmonary fibrosis associated with systemic sclerosis, idiopathic pulmonary fibrosis patients and healthy controls.
Calgranulin B in BAL of patients with IPF and not in other ILD can be explained by massive activation of neutrophils and macrophages subsets at alveolar level in IPF. These cells also contribute to lung fibrosis by releasing different mediators of oxidative stress responsible for tissue damage. Specific interactions of this protein with the endothelial surface suggest a regulatory role in the adhesion of phagocytes to the endothelial surface [11].
Increased levels of calgranulin B have also been reported in other chronic inflammatory disorders (such as cystic fibrosis and COPD) while increases in calgranulin A have been reported in acute inflammation (including ARDS) [12–16]. As IPF is a typical chronic disorder, the increased percentage volumes of calgranulin B found by us in this disease are in line with data in the literature [4]. Our next step will therefore be to perform a full quantitative test such as ELISA, in a larger population of ILD patients to validate these findings.
In our study we failed to find any correlation between calgranulin B percentage volumes in patients and radiological, functional or immunological features; this may be due to the small number of patients analysed in the proteomic study. Quantitative analysis of a larger population using an easier method will now be required to determine whether calgranulin B concentrations in BAL could be a good marker of disease severity.
In conclusion, because calgranulin B, a protein involved in chronic inflammation, has increased expression in IPF and not in other ILDs, it could play a role worthy of further research in the fibrotic processes of IPF.
Abbreviations
- BAL:
-
bronchoalveolar lavage
- ILD:
-
interstitial lung diseases
- 2-DE:
-
two dimensional electrophoresis
References
Foell, D., H. Wittkowski, T. Vogl, and J. Roth. 2006. S100 proteins expressed in phagocytes: a novel group of damage-associated molecular pattern molecules. J. Leukoc. Biol. 81:28–37. doi:10.1189/jlb.0306170.
Lagasse, E., and R. C. Clerc. 1988. Cloning and expression of two human genes encoding calcium-binding proteins that are regulated during myeloid differentiation. Mol. Cell. Biol. 8:2402–10.
Roth, J., S. Teigelkamp, et al. 1992. Complex pattern of myelo-monocytic differentiation antigens MRP8 and MRP14 during chronic airway inflammation. Immunobiology 186:304–14.
Lorenz, E., M. S. Muhlebach, P. A. Tessier, et al. 2008. Different expression ratio of S100A8/A9 and S100A12 in acute and chronic lung diseases. Respir. Med. 102:567–573.
Magi, B., L. Bini, M. G. Perari, et al. 2002. Bronchoalveolar lavage fluid protein composition in patients with sarcoidosis and idiopathic pulmonary fibrosis: a two dimensional electrophoretic study. Electrophoresis 23:3434–44. doi:10.1002/1522-2683(200210)23:19<3434::AID-ELPS3434>3.0.CO;2-R.
Hunninghake, G. W., U. Costabel, M. Ando, et al. 1999. ATS/ERS/WASOG statement on sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 16:149–73.
Meltzer, E. B., and P. W. Noble. 2008. Idiopathic pulmonary fibrosis. Orphanet J Rare Dis. 3:8. doi:10.1186/1750-1172-3-8.
Rottoli, P., B. Magi, M. G. Perari, S. Liberatori, et al. 2005. Cytokine profile and protome analysis in bronchoalveolar lavage of patients with sarcoidosis, pulmonary fibrosis associated with systemic sclerosis and idiopathic pulmonary fibrosis. Proteomics 5:1423–30. doi:10.1002/pmic.200301007.
Bjellqvist, B., C. Pasquali, F. Ravier, et al. 1993. A nonlinear wide-range immobilized pH gradient for two-dimensional electrophoresis and its definition in a relevant pH scale. Electrophoresis 14:1357–65. doi:10.1002/elps.11501401209.
Gorg, A., W. Postel, and S. Gunter. 1988. Two-dimensional electrophoresis with immobilized pH gradients of leaf proteins from barley (Hordeum vulgare): method, reproducibility and genetic aspects. Electrophoresis 9:531–46. doi:10.1002/elps.1150090913.
De Torre, C., S. X. Ying, P. J. Munson, et al. 2006. Proteomic analysis of inflammatory biomarkers in BAL. Proteomics 6:3949–57. doi:10.1002/pmic.200500693.
Merkel, D., W. Rist, P. Seither, et al. 2005. Proteomic study of human bronchoalveolar lavage fluids from smokers with chronic obstructive pulmonary disease by combining surface-enhanced laser desorption/ionization-mass spectrometry profiling with mass spectrometric protein identification. Proteomics 5:2972–80. doi:10.1002/pmic.200401180.
Pedersen, S. K., A. J. Sloane, S. S. Prasad, et al. 2005. An immunoproteomic approach for identification of clinical biomarkers for monitoring disease: application to cystic fibrosis. Mol Cell Proteomics 4:1052–60. doi:10.1074/mcp.M400175-MCP200.
Bowler, R. P., B. Duda, E. D. Chan, and J. J. Enghild. 2004. Proteomic analysis of pulmonary edema fluid and plasma in patients with acute lung injury. Am J Physiol Lung Cell Mol Physiol. 286:L1095–104. doi:10.1152/ajplung.00304.2003.
Kheradmand, F., and D. B. Corry. 2008. Discovery of novel markers in allergic lung inflammation through proteomic-based technologies. Expert Rev Proteomics 5:9–12. doi:10.1586/14789450.5.1.9.
Wattiez, R., and P. Falmagne. 2005. Proteomics of bronchoalveolar lavage fluid. J Chromatogr B Analyt Technol Biomed Life Sci. 815:169–78. doi:10.1016/j.jchromb.2004.10.029.
Acknowledgement
Research financed by MIUR and PAR Siena University.
There is not potential conflict of interest related to the article or the research described.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bargagli, E., Olivieri, C., Prasse, A. et al. Calgranulin B (S100A9) Levels in Bronchoalveolar Lavage Fluid of Patients with Interstitial Lung Diseases. Inflammation 31, 351–354 (2008). https://doi.org/10.1007/s10753-008-9085-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-008-9085-z