
Abstract
The genetic structure of European mudminnow populations throughout the species range was examined using mitochondrial DNA and seven microsatellite loci. Ten mitochondrial haplotypes were detected, suggesting three phylogeographic lineages, which likely diverged during the Early and Middle Pleistocene. These three lineages geographically correspond to three regions: the Danube drainage including the Drava system and Dniester Delta, the Sava system and the Tisza system. High genetic diversity observed using mtDNA was confirmed with microsatellite data, suggesting the existence of 14 populations in the studied area. The isolation-with-migration model showed that migration rates between populations were generally low and were highest between the Drava and its tributary Mura. According to the inferred relative population splitting times, Umbra krameri likely spread from the eastern part of the species range to the west, which also showed the highest genetic diversity and largest population size. As reported by the time-calibrated phylogeny, separation of the European and American Umbra occurred roughly at the end of Late Cretaceous and in the first half of the Paleogene (60.57 Ma with 95% highest probability density of 39.57–81.75). Taking these results into account, appropriate guidelines are proposed to conserve European mudminnow populations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Longitudinal and transverse damming of rivers has altered water flows and the habitats of many freshwater fish species, and is often considered a major cause of the freshwater fish biodiversity crisis (Abell, 2002; Cambray, 1997). Transverse damming is performed for power generation and to improve navigation conditions, whereas longitudinal damming is usually tied to flood control and re-claiming arable land. In lowland landscapes, the predominantly longitudinal damming of rivers has intersected formerly vast wetlands and marshes associated with larger rivers, dramatically altering the landscape. Wetland- and marsh-adapted species have been particularly affected by these changes, and previously vast habitats have been largely diminished, leading to dramatic population declines and local extinctions (Olden, 2016).
A notable example is seen in the European mudminnow (Umbra krameri Walbaum, 1792), a fish specifically adapted to the margins of lowland rivers, floodplains and marshes (Bănărescu et al., 1995; Wanzenböck, 1995, 2004; Bănărescu & Bănăduc, 2007; Pekárik et al., 2014; ). U. krameri could serve as a focal species for this specific type of threatened ecosystem (Lambeck, 1997; Mace et al., 2007) or as indicator species for ecosystem function (Wanzenböck, 2004). It has a relatively small distribution area restricted to the lowlands of the Danube and Dniester drainages. Its populations are believed to be declining in many countries (Mikschi & Wanzenböck, 1995; Bănăduc, 2008); however, some previously unrecorded populations have recently been reported (Trombitsky et al., 2001; Velkov et al., 2004; Govedič, 2010; Sekulić et al., 2013; unpublished data). They are located scattered in small floodplain pools, oxbow lakes and marshy wetlands. During floods, populations may be interconnected, allowing for genetic exchange in a metapopulation framework (Akcakaya et al., 2007).
Umbra krameri has been listed as a vulnerable (VU) species by the IUCN (Freyhof, 2013) and is strictly protected under the Bern convention (Appendix II), and protected by the EU Habitats Directive 92/43/EEC (listed in Annex II) and the national legislation in most countries in its range. While legal protection is guided internationally, conservation efforts are generally localized and are often not coordinated at the national level. Ideally, conservation planning should be performed irrespective of social and administrative entities (i.e. countries or provinces), and should address biologically meaningful spatial scales. This starts with the strategic consideration of conservation management at the spatial scale of the total distribution range and should be successively broken down into smaller spatial scales such as river basins, sub-basins and river stretches. Particularly rare genotypes may deserve higher conservation priority overriding other spatial considerations (Moritz et al., 2002).
Along with U. krameri, the family Umbridae includes four other species according to the traditional taxonomy (Umbridae sensu lato): the central mudminnow (U. limi), eastern mudminnow (U. pygmaea), Olympic mudminnow (Novumbra hubbsi) and Alaska blackfish (Dallia pectoralis), all populating North America, with the latter also extending into northeast Siberia (Kuehne & Olden, 2014).
Genetic studies of the Umbridae are relatively scarce; molecular phylogeny of the family has been primarily studied in the context of higher evolutionary ranks regarding Esociformes and Salmoniformes (e.g. López et al., 2000; López et al., 2004; Campbell et al., 2013; Shedko et al., 2013). These studies revealed that North American and European representatives of Umbra (U. limi, U. pygmaea and U. krameri) are monophlyetic (1), while Umbra, Dallia and Novumbra form a paraphyletic group (2), where Dallia and Novumbra are actually in monophyly with Esox (3). Therefore, it was suggested that the family Umbridae should only contain the genus Umbra and the closely related fossil genera Boltyshia, Paleoesox and Proumbra (Umbridae sensu Gaudant, 2012), while the family Esocidae should also contain Dallia and Novumbra along the genus Esox (Campbell et al., 2013).
Phylogeographic and/or population genetic studies have been performed on Dallia (Campbell & López, 2014; Campbell et al., 2014), N. hubbsi (Pickens, 2003; Adams et al., 2013; DeHaan et al., 2014) and on U. krameri at a limited scale in Serbia, Bosnia-Herzegovina and Hungary (Marić et al., 2015; Takács et al., 2015). Contrary to N. hubbsi, which was found to be genetically homogeneous at the mitochondrial DNA level, Dallia and U. krameri populations showed extensive phylogeographic structuring.
In this study, we analyses the genetic structure of U. krameri using mitochondrial (cytochrome b) and nuclear (tetranucleotide microsatellites) markers (Winkler & Weiss, 2009) throughout most of its range—the Danubian drainage including its major systems, the Drava, Tisza and Sava Rivers, and the Dniester River Delta. This analysis aims to give a comprehensive overview of the phylogeography and population genetics of the species, while also providing fundamental guidelines for its conservation.
In previous phylogenetic studies of the genus Umbra, all three species were not studied together to produce a time-calibrated phylogeny. For that reason, the evolutionary relationship of these three taxa was also examined to infer a time-calibrated phylogeny for the family Umbridae.
Materials and methods
Sampling and DNA isolation
A total of 341 specimens were collected using electrofishing and wattle baskets (Sekulić et al., 2013) from 17 locations across the Danube drainage and the Dniester Delta (Fig. 1; Table 1), from 2011 to 2015. Fin clips were sampled and stored in 96% ethanol. Total DNA was isolated using the phenol–chloroform–isoamyl alcohol method (Sambrook et al., 1989) or DNeasy Blood and Tissue kit (Qiagen, Germany) as per manufacturer instructions. Samples from the five Hungarian populations (Sződ-Rákos-patak, Kolon-tavi-övcsatorna, Ricsei-csatorna, Hejő and Zala) were previously used in a population genetics study of U. krameri in the Hungarian part of the Carpathian Basin (Takács et al., 2015), while two specimens from the Lower Sava (Gromiželj and Bakreni Batar) and one from the Middle Danube (Lugomir) were previously used in a study on the genetic and morphological variability of U. krameri in Serbia and Bosnia and Herzegovina (Marić et al., 2015).

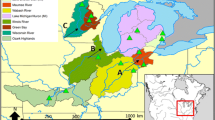
Main diagram: Map of sampling locations. Names and codes of sampling locations are reported in Table 1, with square pie charts representing the distribution and frequencies of mtDNA haplotypes. The Danube and the Dniester drainage area are delineated with thick dotted lines, while the borders between subdrainages are shown with thin dotted lines. The Drava and the Lake Balaton subdrainages are joined together as they were connected and formed a single drainage until the Late Pleistocene. Lower left: The genealogical haplotype network of European mudminnow. Haplotypes are connected with lines that, regardless of length, represent a single mutation. Black circles represent missing or theoretical haplotypes. Haplotype colours correspond to the square pie charts in the central diagram
Mitochondrial DNA
The mitochondrial protein-coding gene cytochrome b (entire length of 1141 bp) was PCR-amplified in 182 individuals (Table 1) using GluF and ThrR primers and the PCR conditions as described in Machordom & Doadrio (2001). Both-directional sequencing was carried out on an ABI Prism 3130xl DNA sequencer using the same primers. DNA sequences were edited and aligned using the programs Chromas Lite 2.01 (http://www.technelysium.com.au/chromas.html; Technelysium Pty Ltd, Australia) and Clustal X (Thompson et al., 1997). Mean genetic net-distances between phyletic lineages were calculated in the program MEGA 5 (Tamura et al., 2011), using the Kimura two-parameter model (Kimura, 1980). The cytochrome b sequences obtained in this study were deposited in GenBank (accession numbers KP898868–KP898876 and KU674836). The genealogical relations between haplotypes were presented as a minimum spanning network (MSN) using the 95% statistical parsimony criterion in the TCS 1.2 program (Clement et al., 2000). Phylogenetic and molecular clock analysis were performed in BEAST v 1.8.3 (Drummond et al., 2012) using the birth–death process (Gernhard, 2008), uncorrelated lognormal relaxed clock (Drummond et al., 2006), TN93 substitution model (Tamura & Nei, 1993), gamma-shaped rate variation (Yang, 1993) and run online on the CIPRES Science Gateway portal v. 3.3 (Miller et al., 2010); the substitution model was selected with ModelGenerator (Keane et al., 2006) based on the Aikake Information Criterion (AIC; Akaike, 1974).
For molecular clock analysis, the dataset was expanded with cyt b sequences of other Esociformes deposited in GenBank (Table 2). As no appropriate calibration dates are available for the Umbra genus, fossil records of other Esociformes were used to calibrate the molecular clock. For the minimum age of all Esociformes, Esteesox, a stem esociform from the late Cretaceous (85 Ma, Wilson et al., 1992) was used, while Esox kronneri Grande, 1999, the first record of the subgenus Kenoza from the late early Eocene (42 Ma, Grande, 1999) was used as a minimum bound for the divergence between Esox lucius and E. niger. For both fossil calibrations, the lognormal priors recommended by Campbell et al. (2013) were applied: the prior for all Esociformes had an offset of 85.0 with a mean of 1.0 and a SD of 1.0 (5% CI 85.5, 95% CI 99.1), and the prior for the split within Esox had an offset of 42.0 with a mean of 1.0 and SD 0.65 (5% CI 45.9, 95% CI 52.9). Calculations were conducted using the BEAGLE library (Ayres et al., 2012) and run in three independent runs of 30,000,000 generations sampled every 3000 generations. After verifying adequate sampling (ESS > 200) and convergence with Tracer (Rambaut et al., 2014), a 10% burn-in was applied and the tree files were combined with LogCombiner. Finally, TreeAnnotator was used to calculate a maximum clade credibility tree, median values of divergence times, posterior probabilities and bounds for the 95% highest posterior density (HPD) interval.
Microsatellites
Seven microsatellite loci (UkrTet1, UkrTet3–UkrTet8) were amplified in 341 individuals (Table 1), according to previously published protocols (Winkler & Weiss, 2009). Fragment analysis was performed on a 3130xl Genetic Analyzer and genotyped using Gene-Mapper v4.0 (Applied Biosystems).
The presence of null alleles, gene diversity (heterozygosity), F statistics, as well as inter-population allele sharing distances (D AS), were calculated for all populations using Microchecker v2.2.3 (Van Oosterhout et al., 2004), GENETIX 4.04 (Belkhir et al., 1996–2004), FSTAT 2.9.3.2 (Goudet, 2002) and Populations software (Langella, 2002), respectively. Private alleles and allelic richness were estimated by rarefaction analysis, using ADZE (Szpiech et al., 2008), to compare genetic diversity among populations despite unequal sample numbers and to assess whether sampling effort was sufficient to capture genetic diversity. To determine whether stepwise-like mutations have contributed to genetic differentiation (Hardy et al., 2003), allele size (R ST) and the allele identity—based measure (F ST) were compared by testing whether the observed R ST was larger than the value obtained after permuting allele sizes among alleles within populations (pR ST) as implemented in SPAGeDI 1.3 (Hardy & Vekemans, 2002; 20,000 permutations).
Genetic differentiation of the whole sample set was assessed using hierarchical STRUCTURE analysis (Pritchard et al., 2000; Vähä et al., 2007). STRUCTURE 2.3.2.1 runs Markov chain Monte Carlo (MCMC) simulations to partition individuals into K clusters. For runs estimating ln Pr(X|K) under a certain K, different run lengths were used (from 20,000 to 100,000 burn-in and 100,000 to 500,000 total length, repeated seven times for each K) depending on convergence. Stepwise exclusion of the most differentiated clusters was conducted in the hierarchical STRUCTURE analysis, allowing for more precise clustering of the remaining individuals without eliminating admixed individuals. Each excluded cluster was investigated for possible hidden substructures by choosing K values according to each specific setting (Vähä et al., 2007). The ΔK method (Evanno et al., 2005) was applied to estimate the most probable K (Appendix 1 in Supplementary Material).
In order to determine the amount of gene flow between populations and to estimate the parameter theta (θ), the isolation-with-migration model (IM, as implemented in the software IMa2; Hey & Nielsen, 2007) was used. Mitochondrial cytochrome b gene and microsatellite loci were analysed together. The HKY model of sequence evolution was applied to mitochondrial sequences, and a stepwise mutation model (SMM) was assumed for microsatellite loci. IM was applied to all neighbouring populations and to several additional combinations selected on the basis of the results from the STRUCTURE analysis (total of 23 combinations; all tested populations are listed in Appendices 2, 3 and 4 in the Supplementary Material). Five parameters were estimated for each combination: current and ancestral population sizes (θ 1, θ 2 and θ ANC, respectively), relative time since divergence (t) and a single migration parameter (m). Upper bounds for parameter priors were estimated for each tested population pair from consecutive preliminary runs of the program, based on initial estimates of θ as advised in the IMa2 manual and span: −q (30–400), −m (0.75–4) and −t (4–30). In all combinations, 100 Markov chains were run in parallel under a geometric heating scheme. Several shorter trial runs with different heating schemes were explored between selection of population pairs (6) to identify high swapping rates between adjunct chains. The settings (ha = 0.99, hb = 0.3) gave adequate swapping rates (40–80% between the majority of adjunct chains) and resulted in good mixing of the Markov chains for all tested population pairs. For the final simulations, the manual recommendations for large datasets were followed. For each tested combination, two independent jobs were run until a suitable burn-in was reached for at least 1,000,000 steps. Next, a new set of runs was started by reloading the Markov chain state file with an additional short burn-in period of 100,000 steps and afterwards 20,000 genealogies were sampled every 50 steps from a total 1,000,000 steps. Both Markov chain state files generated in burn-in runs were used two times. Finally, all four replicates were combined in L-mode run with identical parameter settings. Based on the 1.3 ± 0.5 average age of maturity (Kuehne & Olden, 2014), migration events were assessed using a generation time of two years.
Results
MtDNA analysis
Aligned sequences of the 1085 bp 3′-end cytochrome b mtDNA gene obtained from 182 individuals grouped into ten haplotypes: Da1 and Sa1 were previously identified in the Danube and Sava drainages in Serbia and Bosnia-Herzegovina (Marić et al., 2015), while the remaining haplotypes (i.e. Da2, Da3, Da4, Da5, Da6, Da7, Ti1 and Ti2) were not previously described. The haplotypes Da1, Da2 and Da3 were found predominantly in the Upper and Middle Danube, while Da4 and Da5 were detected only in the Danube Delta. Da6, the most frequent and widespread haplotype was restricted to the Drava River system, Lake Balaton, the Lower Danube River and the Dniester Delta, but was completely lacking in the Upper and Middle Danube. Da7 was detected only in the Dniester Delta, Ti1 and Ti2 only in the Tisza River system in Hungary. Sa1 was detected in the Sava River system and in the Danube River, though only in proximity to the Sava mouth (Table 1).
The phylogenetic reconstruction of the mitochondrial haplotypes as inferred from the Bayesian tree (Fig. 2) supports the monophyly of the Umbra genus and the sister relationship between U. limi and U pygmaea (López et al., 2000, 2004). U. krameri clustered into two clades: one was statistically well supported (0.97 posterior probability) and comprised very similar haplotypes found in the Drava, Balaton, Danube and Dniester locations (Danube phyletic lineage), while the second showed only weak support (0.47 posterior probability). This less supported clade is formed by two phyletic lineages, represented by two haplotypes detected only in the Tisza River (Tisza phyletic lineage), and a haplotype detected primarily in the Sava River sites (Sava phyletic lineage). Mean net-distances between the three lineages were 0.003 between the Danube and Tisza phyletic lineages, 0.006 between the Danube and the Sava phyletic lineages, and 0.005 between the Tisza and the Sava phyletic lineages.

Fossil calibrated phylogeny of Esociformes generated using a relaxed clock in BEAST. 95% HPD intervals are shown as grey bars at nodes. Median node ages are shown as node labels. The upper left square shows the same Bayesian phylogenetic tree with branch lengths representing substitutions, the scale bar indicates the number of substitutions per site, and posterior probabilities are shown as node labels
MSN (Fig. 1) supported the topology represented by the Bayesian tree (Fig. 2) and revealed the central position of the haplotype Da6 as being one or two mutation steps from the other Da haplotypes, four mutations from the Ti1 and Ti2 haplotypes, and six from the Sa1 haplotype.
The molecular clock analysis based on the alignment of ten U. krameri haplotypes and six other Umbirdae and Esocidae species with two calibration points (Esteesox for Esociformes and E. kronneri for the first record of the subgenus Kenoza) yielded a divergence time for the European and American Umbra species of 60.57 Ma (with a 95% highest probability density (HPD) of 39.57–81.75 Ma), while the diversification within U. krameri was estimated to start at 1.01 Ma (with 95% HPD of 0.48–1.74) (Fig. 2). The Tisza + Sava lineages first separated from the Danubian ones, followed by the splitting of the Sava and Tisza lineages at 0.70 Ma (with 95% HPD of 0.19–0.90).
Microsatellite DNA analysis
Rarefaction analysis revealed allelic richness that varied from 3.4 to 11.1, and observed heterozygosity varied from 0.331 to 0.819, with the highest values observed in Enisala (Danube Delta) and the Dniester Delta, and the lowest in Šuma Žutica (Sava) (Table 1). Rarefaction analysis showed that the private allelic richness in populations from both deltas (0.64 in the Danube and 1.63 in the Dniester) did not decrease with an increasing number of individuals (Fig. 3a; Table 1). Furthermore, certain population group combinations (between Lower Danube & Dniester, Upper & Middle Danube, Drava & Balaton, Sava and Tisza; Table 1) showed that the Lower Danube & Dniester–Tisza combination exhibited the highest private allele sharing, closely followed by the combinations Sava–Lower Danube & Dniester, Lower Danube & Dniester–Drava & Balaton, and Upper & Middle Danube–Lower Danube & Dniester (Fig. 3b). Neither null alleles nor deviations from Hardy–Weinberg equilibrium were detected in the examined populations.

Rarefaction analysis of private alleles for five major clusters as inferred from geography and population analysis (a), and rarefaction analysis of shared private alleles for combinations of major population clusters (b). Upper–Middle Danube (locations 1–6; Table 1), Lower Danube–Lower Dniester (locations 7, 8 and 17), Tisza, Sava including Kraljevac in the Middle Danube, and Drava–Balaton including Mura
The degree of differentiation among the 17 analysed populations was significant and relatively high in most cases and spanned from 0.022–0.514 for pairwise cnd 0.108–0.915 for D AS (Table 3).
In the hierarchical STRUCTURE analysis, the most probable numbers of K values were K = 2 for the 1st and 3rd steps and K = 5 for both 2nd steps (for details see Appendix 1 in Supplementary Material). In accordance with these K values, European mudminnows were partitioned into two groups in the first step: the Upper and Middle Danube (Lugomir and upstream locations), Mura, Drava and Balaton (group I), and the Sava, Tisza, Middle and Lower Danube (Kraljevac and downstream locations) and Dniester Delta, (group II). In further steps, additional partitioning within both groups became evident; in group I in the Upper and Middle Danube, each sampling site represented a genetically well-defined homogeneous population. The Drava (Županijski kanal) and Balaton (Zala) population showed some inter-population genetic mixing with the genetically similar population from the Mura River (Nagy Parlag). In group II, Comana (Lower Danube) and Šuma Žutica (Sava) formed distinct and well-defined homogeneous populations. Populations from the Lower Sava (Bakreni Batar and Gromiželj) and from the Middle Danube near the mouth of the Sava (Kraljevac) represented an admixture of two distinct genetic units. Within the group of remaining locations, Palanca–Mayaki (Dniester) and Ricsei-csatorna (Tisza) were genetically homogeneous and distinctive, while individuals from the Hejő (Tisza) and Enisala (Danube Delta) exhibited admixed genotypes derived from the previous two populations. Further intra-population partitioning did not reveal any additional clusters (Fig. 4; Appendix 1 in Supplementary Material).

Estimated population structure as inferred by hierarchical STRUCTURE analysis of microsatellite marker DNA data. Black lines separate sampling sites. After three steps, 14 clusters were identified. The most probable K for the analysed samples shown in the arrows is based on the ΔK method; no further structures were detected in subsequent rounds (after the third step) and within the excluded clusters (K = 1). Arrows delineate the progress of the hierarchical approach, where subsets of the data were subsequently analysed
The observed R ST value of the whole sample set was 0.436, while the pR ST value was 0.151 (P = 0.0000) and the F ST value was 0.207. The significantly higher R ST than pR ST, and considerably higher value than F ST, suggested that SMM contributed to genetic differentiation; furthermore, no non-tetra nucleotide repeat motifs were observed. IM analysis of neighbouring populations revealed a stepping-stone pattern with low levels of migration (Fig. 5; Appendixs 3 and 4 in Supplementary material). In the majority of tested population pairs, runs produced clear peaks and replicates resulted in similar estimates of all parameters. An arbitrary value of 0.05 for the bin with the highest value in the migration histogram (HiPt) was used to identify migration rates greater than zero. The strongest migration paths were observed between Mura (Nagy Parlag) and Drava (Županijski kanal) and between the Middle Danube (Kraljevac) and Lower Sava (Gromiželj and Bakreni Batar joined). When converting the migration parameter into per-generation population migration rates (M = θ × m/2), peak locations corresponded to 2.61 (M MUR → M DRA) and 0.56 (M DRA → M MUR) migration events per generation between Mura and Drava and to 1.12 (M M.DAN → M L.SAV) and 3.35 (M L.SAV → M M.DAN) events between the Middle Danube and Lower Sava. This suggests 1.31 or 0.28 migration events per year between the Mura and Drava and 0.56 or 1.68 events between the Middle Danube and Lower Sava when taking into account the average European mudminnow generation time (two years). Within the Danubian watershed, migrations were generally higher in the Middle and Upper Danube and in the Sava, Tisza and Drava-Balaton watersheds than in the Lower Danube. Only negligible migration was detected between Comana and Enisala, while migration rate between the Lower (Comana) and the Middle Danube (Kraljevac) was very small. The IM model also revealed no trans-watershed migrations between the Sava and Drava and Middle Danube and Tisza, while low levels of migrations were detected between the Danube Delta (Enisala) and the Upper Tisza (Ricsei-csatorna). Migration rates between the Danube Delta and the Upper Tisza correspond to 0.09 (M D.DAN → M U.TIS) and 1.83 (M U.TIS → M D.DAN) migration events per year.

Migration patterns according to IM model estimates. Arrow width corresponds to IMa2’s HiPt estimate of migration rate as presented in the legend (upper left). Only migration rates above 0.05 are shown; see Appendix 3 in Supplementary Material and Table 1 for names of sampling locations
The IM model showed that Enisala (Danube Delta) and Palanca–Mayaki (Dniester Delta) populations were the largest, with θ values of 44.78 and 25.10, respectively, followed by the Hejő (Middle Tisza; θ = 12.09) and Kraljevac (Middle Danube; θ = 8.37), while Šuma Žutica (Middle Sava) was the smallest population with the lowest theta value (θ = 0.40). In the Županijski kanal (Drava; θ = 5.87), considerable variation was observed in the θ estimation between the tested population pairs (Table 1 and Appendix 2 in Supplementary Material). In addition, the relative times since divergence calculated by the IM model were generally older in the eastern range of the species, especially when comparing the Enisala population (Danube Delta) to other populations (Appendix 3 in Supplementary Material).
Discussion
Phylogeography and molecular clock analysis of U. krameri
The phylogenetic mtDNA analysis of the extensive sample set, which covered the majority of the U. krameri range, revealed three phyletic lineages that corresponded closely to three main rivers in the area: Danube, Sava and Tisza.
Diversification within U. krameri started at approximately 1.01 Ma (0.48–1.74), which is in general agreement with the time frame set by Marić et al. (2015), who proposed that the Sava lineage separated from the Danubian lineage approximately 0.70 Ma ago. The time span of the presumed diversification of the species includes two Pleistocene glaciations (Gűnz and the first phase of Mindel) (Penck & Brückner, 1909; Gibbard & van Kolfschoten, 2004). Although the areas inhabited by these mudminnow populations were not covered with ice sheets (Mangerud et al., 2004), indirect effects of glacial events could have shaped the river network of the middle Danube. Furthermore, intensive tectonic movements occurred in the same period, which may have resulted in significant shifts of river courses within the basin (Brilly, 2010). Thus, the palaeogeological events of the Early and Middle Pleistocene could have played a significant role in shaping the genetic differentiation of U. krameri, likely separating the Sava and the Tisza populations from the Danube–Mura-Drava–Balaton populations, and initiating their genetic divergence.
As mudminnow can thrive only in a narrow range of environmental conditions and is sensitive to competition, in addition to major Pleistocene geological events, even simple random habitat fragmentation may have led to population isolation and lineage formation.
The most frequently observed haplotype (Da6) was also the most widespread and was found in the most distant sites of the species range, i.e. in the Mura River in the west and the Dniester Delta in the east. Also, Da6 appeared to be the central and presumably the ancestral haplotype of the entire species. Yet, its modern distribution is patchy; its occurrence in the Drava River and relative proximity to Lake Balaton is expected and likely reflects Late Pleistocene communication between the two systems, as proposed by Gábris & Mari (2007). The question arises as to why this haplotype is lacking in the Upper and Middle Danube, where it is substituted with its derivatives (Da1–Da3) and why it is again abundant in the Lower Danube and Dniester Delta. Considering that U. krameri is a habitat specialist, and also that mtDNA has a higher level of genetic drift than nuclear DNA markers, the most likely explanation for the patchy distribution of Da6 haplotype appears to be genetic drift.
Time-calibrated phylogeny for Umbridae family
This phylogenetic analysis confirmed the paraphyly of the Umbridae family (Umbra, Novumbra, Dallia), placing Novumbra and Dallia within the Esocidae (Gaudant, 2012; Campbell et al., 2013; Shedko et al., 2013) and confirming the monophyly of the genus Umbra (López et al., 2000, 2004). Previous phylogenetic studies of the Umbridae family did not examine all three Umbra species together to produce a time-calibrated phylogeny (c.f. López et al., 2000, 2004; Campbell et al., 2013; Shedko et al., 2013), and thus the time of the split between the North American and European Umbra was not resolved. According to the time-calibrated phylogeny presented here, Umbra separated from the rest of the Esociformes approximately at the end of the Early or in the Late Cretaceous, which is comparable to the time estimation in Campbell et al. (2013), while the separation of the European and American Umbra species roughly spans the end of the Late Cretaceous into the first half of the Paleogene. During that period, the Atlantic Ocean was already well formed, separating Eurasia and North America (Scotese, 2001), and thus this estimate places the split between the European and American Umbra much later than at the breakup of the Laurasian supercontinent. The molecular results presented here indicate that the split between the North American and European Umbra pre-dates the oldest known fossil representative of the genus Umbra, collected in Northern Bohemia and dating to the Late Oligocene (U. prochazkai Oberhlová, 1978), making ancestral Umbra a contemporary of the oldest known fossil representative of the Umbridae family (sensu Gaudant, 2012) collected in the Boltyshka basin of Ukraine and dating to the late Palaeocene (Boltyshia brevicauda Sytchevskaya & Daniltschenko, 1975). The split between the North American and European Umbra is decidedly deeper than the split between the subgenera Kenoza and Esox and is comparable to the split between the genera Novumbra and Esox in terms of the molecular clock analysis. Therefore, differences between the North American and European Umbra could well be interpreted at the genus level; or at least, classification into the subgenus Melanura (Agassiz, 1853) as defined by Nelson (1972) should be followed.
Based on the time-calibrated phylogeny presented in this study, the distribution of ancestral Umbra might have extended bi-continentally either across the North Atlantic Land Bridge (NALB) and/or the Beringia Land Bridge (BLB), which linked the continents across the Atlantic and the Pacific oceans and were available intermittently from the beginning of the Paleocene (Brikiatis, 2014), with final subsidence of the NALB during the late Miocene (Tiffney, 1985; Denk et al., 2011) and loss of the BLB near the end of the Pleistocene (Gladenkov et al., 2002). However, given that the native distribution of Umbra in North America (subgenus Melanura) is exclusive to the Atlantic drainage, and that the distribution of Umbra in Eurasia (subgenus Umbra) is restricted to Central Europe and the Black Sea watershed (including the fossil record), the distribution of a once common ancestor most likely extended across the North Atlantic Land Bridge. Furthermore, a lack of fossils from the family Umbridae (sensu Gaudant, 2012) from North America indicates that the genus may have originated in Europe. A similar biogeographic origin and distribution pattern between sister lineages was recently described for the freshwater fish genera Sander (Haponski & Stepien, 2013) and Perca (Stepien et al. 2015), where North American and European sister lineages diverged much later than the breakup of Laurasia and coincided with the closure of the NALB during the Miocene. The examples of Sander and Perca clearly demonstrate that such biogeographic scenarios are also possible with freshwater fish.
Population genetics and demography of U. krameri
Although the results of the mtDNA analysis of U. krameri showed a considerable level of genetic variation observable through the clustering of haplotypes into three phylogeographic lineages, analysis of microsatellite loci allowed for a more precise resolution of genetic variation. Namely, the pairwise FST values (Table 3) revealed a strongly significant statistical difference between 16 of the 17 sampled locations, with the exception of Bakreni Batar and Gromiželj (Lower Sava), where mudminnows were recognized as a uniform population. In addition to this pair, STRUCTURE analysis did not separate populations from Županijski kanal (Drava)–Zala (Lake Balaton) and Enisala (Danube Delta)–Hejő (Lower Tisza). Therefore, the Danube watershed and Dniester Delta harbour at least 14 genetically differentiated populations of U. krameri.
Regarding the population pairs from the Lower Sava (Bakreni Batar–Gromiželj) and from the Drava and Lake Balaton (Županijski kanal–Zala), the genotype clustering results are not surprising, as respective pairs were physically connected until recently (Gábris & Mari, 2007; Marić et al., 2015). This is also supported by the shared haplotypes (Sa1 and Da6, respectively) (Table 1). However, the relationship between the two deltas (especially Danube Delta) and the Middle Tisza population remains puzzling. Although these three populations are geographically very distant, and though the populations from both deltas share no haplotypes with the Tisza population, microsatellite analysis suggested their similarity. But even so, the IM model showed no migration between them, which suggests that the apparent genetic similarity is likely a consequence of ancestral polymorphism rather than gene flow. This assumption is congruent with the fact that ancestral alleles persist longer in large populations, such as Enisala, Palanca–Mayaki and Hejő, the three largest populations in this study. Furthermore, large effective population sizes can lead also to allelic saturation. If so, similar allelic frequency profiles may not indicate recent extensive genetic exchange or retention of ancestral polymorphisms, but could reflect size homoplasy leading to misinterpretations of long-term relationships (Estoup et al., 2002).
A strong genetic spatial structure is also reflected by the inferred migration pattern. The IM approach detected a stepping-stone migration pattern with low levels of migration. Converted per-generation population migration rates generally correspond to the migration of less than one individual per generation (Appendix 4, in Supplementary Material). In general, migration was higher in the Middle and Upper Danube and in the Sava, Tisza and Drava-Balaton watersheds, and lower in the Lower Danube. As discussed above, the only population where no migration with neighbouring populations was observed was Palanca–Mayaki in the Dniester Delta. This absence of gene flow between the Dniester and the Danube Delta excludes a migration pathway through the Black Sea, a speculation originating from the observation of the species in the Black Sea (Raykov et al., 2012), which was most likely false (Hajdú et al., 2015). In addition, the IM model revealed no trans-watershed migration between the Sava and Drava and Middle Danube and Tisza, while low levels of migration were detected between the Danube Delta (Enisala) and the Upper Tisza (Ricsei-csatorna). Although a connection between these two populations could theoretically be explained by ancestral polymorphism, this is highly unlikely, as the IM approach distinguishes between potential ancestral polymorphism and recurrent contemporary gene flow occurring after population separation (Marko & Hart, 2012). However, genetic similarity due to size homoplasy associated with mutation-driven saturation effects cannot be excluded. Not considering the cluster joining the Danube Delta and Hejő population, the STRUCTURE analysis largely coincided with the results from the IM approach and confirmed higher gene flow within the identified clusters (e.g. between Patašský kanál and Kolon-tavi-övcsatorna within the Drava-Balaton watershed).
Takács et al. (2015) estimated similar but somewhat higher migration rates between mudminnow populations from the Carpathian basin using MIGRATE-N. Direct comparison is difficult, as Takács et al. (2015) pooled their samples according to the STRUCTURE analysis despite large distances between sampling sites. They reported the highest rates (>1.5 individuals per generation) between the Middle Hungarian Region (including Kolon-tavi-övcsatorna) and Hanság-Szigetköz in the Middle Danube, from Balaton to Mura in the Drava-Balaton watershed, and from Middle Tisza including the Köros River watershed to the Upper Tisza. The different migration rates detected between these studies are likely due to the use of different migration estimation software. While MIGRATE-N assumes that the size and the population structure have been stable for ~4 N ef generations, IMa2 does not make this assumption and thus is well suited for the analysis of younger populations (reviewed in Kuhner, 2009). Therefore, when the ratio between N ef and the splitting time is high, MIGRATE-N cannot distinguish between gene flow and shared ancestral polymorphism, leading to an overestimation of migration rates (Marko & Hart, 2012). Furthermore, population subdivision can affect migration rate estimates (Wakeley et al., 2000).
Defining units for conservation purposes
In comparing the genetic diversity of U. krameri (Table 1) and its counterparts D. pectoralis and N. hubbsi in North America (Campbell et al., 2014; DeHaan et al., 2014), the highest allelic richness was detected in the lowest reaches of the largest rivers in all three species. The populations from the Danube and Dniester deltas displayed the highest microsatellite diversity and the largest effective population sizes. Such large differences in θ between these two populations and those from other locations can be attributed to the wide range of habitats in the Danube and Dniester deltas in comparison with upstream locations, e.g. the Danube Delta which covers a vast area of approximately 4152 km2, also had the highest number of detected mtDNA haplotypes (4 of 10; Table 1), with the central Da6 haplotype as dominant (Fig. 1). Furthermore, both deltas and the entire Lower Danube are the only areas where private allelic richness increased with sample size (Fig. 3a). Exceptional parameters of genetic polymorphism and high effective population sizes in both deltas indicate that the eastern part of the species range should be considered the centre of the species diversity. Rich genetic diversity in deltas could be attributed to the stochastic dynamics typical of large populations, where the effects of genetic drift is minor compared to small populations, causing allelic richness to increase with sample size. The population divergences estimated using the IM model showed that the splits between neighbouring populations were oldest in the eastern part of the range. This suggests a possible expansive role of habitats in the delta regions, from where U. krameri likely spread to the west (i.e. to the remaining sampling area) and not vice versa. This is also supported by the shared private alleles (mean number of private alleles for major population cluster combinations) found in the Upper and Middle Danube from the Lower Danube & Dniester and in the Tisza, Sava, Drava & Balaton, (Fig. 3b).
Mitochondrial DNA analysis revealed some genetic divergence among three geographically well-defined groups, the Tisza, Sava and Danube (mean net-distances between them were from 0.003 to 0.006), indicating a certain period of their distinct evolution (see discussion above). This is also supported by the estimation of inter-population gene flow, which was minimal among those three groups, suggesting considerable reproductive isolation. For these reasons, these three phyletic lineages, as defined by the three haplogroups, could be considered potential evolutionary significant units (ESU). On the other hand, the uneven distribution of microsatellite polymorphism among the small sampled populations and high genetic structuring within each of the three phyletic lineages may not reflect a natural evolutionary process but rather random drift governed by recent habitat fragmentation as a result of human impact (e.g. damming). For example, the smallest population with the lowest genetic diversity was detected in an isolated locality in the Sava river system (Šuma Žutica) in Croatia (Table 1) covering just a few square kilometres, with no other known records of U. krameri in the region. Adaptive differentiation seems unlikely in very recently split populations with small Ne, as there is simply no time for selection to take place. In cases like this, it is questionable whether such small populations represent genetically viable entities with a good prospect of long-term survival. Therefore, caution should be taken when delineating ESUs on the basis of microsatellites, as these markers known for their high mutation rate and neutral evolutionary history are likely to result in excessive splitting of populations (Frankham et al., 2012) and are generally inadequate for characterizing adaptive patterns (Funk et al., 2012).
References
Abell, R., 2002. Conservation biology for the biodiversity crisis: a freshwater follow-up. Conservation Biology 16: 1435–1437.
Adams, B., P. W. DeHaan, R. Tabor, B. Thompson & D. K. Hawkins, 2013. Characterization of tetranucleotide microsatellite loci for Olympic mudminnow (Novumbra hubbsi). Conservation Genetics Resources 5: 573–575.
Agassiz, L., 1853. Recent researches of Prof. Agassiz. American Journal of Science and Arts 16: 134–136.
Akaike, H., 1974. A new look at the statistical model identification. IEEE Transactions on Automatic Control 19: 716–723.
Akcakaya, H. R., G. Mills & C. P. Doncaster, 2007. The role of metapopulations in conservation. In Macdonald, D. W. & K. Service (eds), Key Topics in Conservation Biology. Blackwell Publishing, Oxford: 64–84.
Ayres, D. L., A. Darling, D. J. Zwickl, P. Beerli, M. T. Holder, P. O. Lewis, J. P. Huelsenbeck, F. Ronquist, D. L. Swofford, M. P. Cummings, A. Rambaut & M. A. Suchard, 2012. BEAGLE: an application programming interface and high-performance computing library for statistical phylogenetics. Systematic Biology 61: 170–173.
Bănăduc, D., 2008. Umbra krameri Walbaum, 1792 a Natura 2000 protected fish species, in Romania. Acta Ichtiologica Romanica 3: 33–44.
Bănărescu, P. M. & D. Bănăduc, 2007. Habitats Directive (92/43/EEC) fish species (Osteichthyes) on the Romanian territory. Acta Ichtiologica Romanica 2: 43–78.
Bănărescu, P. M., V. Otel & A. Wilhelm, 1995. The present status of Umbra krameri Walbaum in Romania. Annalen des Naturhistorischen Museums in Wien 97B: 496–501.
Belkhir, K., P. Borsa, L. Chikhi, N. Raufaste & F. Bonhomme, 1996–2004. GENETIX v. 4.04, Logiciel sous WindowsTM pour la Ge´ne´tique des Populations. Universite´ Montpellier 2, Laboratoire Ge´nome et Population, Montpellier.
Brilly, M., 2010. Hydrological Processes of the Danube River Basin: Perspectives from the Danubian Countries. Springer, New York.
Brikiatis, L., 2014. The De Geer, Thulean and Beringia routes: key concepts for understanding early Cenozoic biogeography. Journal of Biogeography. doi:10.1111/jbi.12310.
Cambray, J., 1997. Freshwater fish—a global biodiversity crisis. Hydrobiologia 353: 199–202.
Campbell, M. A. & J. A. López, 2014. Mitochondrial phylogeography of a Beringian relict: the endemic freshwater genus of blackfish Dallia (Esociformes). Journal of Fish Biology 84(2): 523–538.
Campbell, M. A., J. A. López, T. Sado & M. Miya, 2013. Pike and salmon as sister taxa: detailed intraclade resolution and divergence time estimation of Esociformes + Salmoniformes based on whole mitochondrial genome sequences. Gene 530: 57–65.
Campbell, M. A., G. K. Sage, R. L. DeWilde, J. A. López & S. L. Talbot, 2014. Development and characterization of 16 polymorphic microsatellite loci for the Alaska blackfish (Esociformes: Dallia pectoralis). Conservation Genetics Resources 6(2): 349–351.
Clement, M., D. Posada & K. Crandall, 2000. TCS: a computer program to estimate gene genealogies. Molecular Ecology 9: 1657–1660.
DeHaan, P. W., B. A. Adams, R. A. Tabor, D. K. Hawkins & B. Thompson, 2014. Historical and contemporary forces shape genetic variation in the Olympic mudminnow (Novumbra hubbsi), an endemic fish from Washington State, USA. Conservation Genetics 15: 1417–1431.
Denk, T., F. Grimsson, R. Zetter & L. A. Simonarson, 2011. The biogeographic history of Iceland—the North Atlantic Land Bridge revisited. Topics in Geobiology 35: 647–668.
Drummond, A. J., S. Y. W. Ho, M. J. Phillips & A. Rambaut, 2006. Relaxed phylogenetics and dating with confidence. PLoS Biology 4: e88. doi:10.1371/journal.pbio.0040088.
Drummond, A. J., M. A. Suchard, D. Xie & A. Rambaut, 2012. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Molecular Biology and Evolution 29: 1969–1973.
Estoup, A., P. Jarne & J.-M. Cournet, 2002. Homoplasy and mutation model at microsatellite loci and their consequences for population genetics analysis. Molecular Ecology 11: 1591–1604.
Evanno, G., S. Regnaut & J. Goudet, 2005. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Molecular Ecology 14: 2611–2620.
Felsenstein, J., 1985. Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39: 783–791.
Frankham, R., J. D. Ballou, M. R. Dudash, M. D. B. Eldridge, C. B. Fenster, R. C. Lacy, J. R. Mendelson, I. J. Porton, K. Ralls & O. A. Ryder, 2012. Implications of different species concepts for conserving biodiversity. Biological Conservation 153: 25–31.
Freyhof, J., 2013. Umbra krameri. The IUCN Red List of Threatened Species. Version 2014.2. www.iucnredlist.org. Accessed on 11 November 2014.
Funk, W. C., J. K. McKay, P. A. Hohenlohe & F. W. Allendorf, 2012. Harnessing genomics for delineating conservation units. Trends in Ecology and Evolution 27(9): 489–496.
Gábris, Gy & L. Mari, 2007. The Pleistocene beheading of the Zala River (West Hungary). Földrajzi értesítő 56: 39–50. [In Hungarian with an English summary].
Gaudant, J., 2012. An attempt at the palaeontological history of the European mudminnows (Pisces, Teleostei, Umbridae). Neues Jahrbuch Fur Geologie Und Palaontologie-Abhandlungen 263(2): 93–109.
Gernhard, T., 2008. The conditioned reconstructed process. Journal Theoretical Biology 253: 769–778.
Gibbard, P. & T. van Kolfschoten, 2004. The Pleistocene and Holocene epochs. In Gradstein, F. M., J. G. Ogg & A. G. Smith (eds), A Geologic Time Scale. Cambridge University Press, Cambridge: 441–452.
Gladenkov, A. Y., A. E. Oleinik, L. Marincovich & K. B. Barinov, 2002. A refined age for the earliest opening of Bering Strait. Palaeogeography Palaeoclimatology Palaeoecology 183: 321–328.
Goudet, J., 2002. FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3.2). http://www2.unil.ch/popgen/softwares/fstat.htm. Accessed 15 October 2009.
Govedič, M., 2010. Ribe reke Drave med Mariborom in Hrvaško mejo 35 let po izgradnji hidroelektrarn [Fish of Drava river between Maribor and Croatian border 35 years after construction of hydro power plants]. In Book of Abstracts–International conference on the Drava River–Life in the river basin, Slovenia, Dravograd (30. 09.–01. 10. 2010.). Jovan Hadži Institute of Biology Scientific Research Centre SASA, Municipality Dravograd, RRA Koroška–Regional Development Agency for Koroška, Ljubljana–Dravograd: 22–23.
Grande, L., 1999. The first Esox (Esocidae: Teleostei) from the Eocene Green River Formation, and a brief review of esocid fishes. Journal of Vertebrate Paleontology 19: 271–292.
Grande, T., H. Laten & J. López, 2004. Phylogenetic relationships of extant Esocid species (Teleostei: Salmoniformes) based on morphological and molecular characters. Copeia, 2004: 743–757.
Hajdú, J., L. Várkonyi, J. Ševc & T. Müller, 2015. Corrective notice to the European mudminnow (Umbra krameri Walbaum, 1792) record from the Black Sea. Biologia 70: 1429–1431.
Haponski, A. E. & C. A. Stepien, 2013. Phylogenetic and biogeographical relationships of the Sander pikeperches (Percidae: Perciformes): patterns across North America and Eurasia. Biological Journal of the Linnean Society 110: 156–179.
Hardy, O. J. & X. Vekemans, 2002. SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Molecular Ecology Notes 2: 618.
Hardy, O. J., N. Charbonnel, H. Freville & M. Heuertz, 2003. Microsatellite allele sizes: a simple test to assess their significance on genetic differentiation. Genetics 163: 1467–1482.
Hey, J. & R. Nielsen, 2007. Integration within the Felsenstein equation for improved Markov Chain Monte Carlo methods in population genetics. Proceedings of the National Academy of Sciences 104: 2785–2790.
Ishiguro N. B., M. Miya & M. Nishida, 2003. Basal euteleostean relationships: a mitogenomic perspective on the phylogenetic reality of the “Protacanthopterygii”. Molecular Phylogenetics and Evolution 27: 476–488.
Keane, T. M., C. J. Creevey, M. M. Pentony, T. J. Naughton & J. O. McInerney, 2006. Assessment of methods for amino acid matrix selection and their use on empirical data shows that ad hoc assumptions for choice of matrix are not justified. BMC Evolutionary Biology 6: 29. doi:10.1186/1471-2148-6-29.
Kimura, M., 1980. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. Journal of Molecular Evolution 16: 111–120.
Kuehne, M. L. & D. J. Olden, 2014. Ecology and conservation of mudminnow species worldwide. Fisheries 39: 341–351.
Kuhner, M. K., 2009. Coalescent genealogy samplers: windows into population history. Trends in Ecology and Evolution 24: 86–93.
Lambeck, R. J., 1997. Focal species: a multi-species umbrella for nature conservation. Conservation Biology 11: 849–856.
Langella, O., 2002. Populations 1.2.28. Logiciel de génétique des populations. Laboratoire Populations, génétique et évolution, CNRS UPR 9034, Gif-sur-Yvette. http://www.cnrs-gif.fr/pge/. Accessed 10 October 2009.
López, A., P. Bentzen & W. Pietsch, 2000. Phylogenetic relationships of Esocoid fishes (Teleostei) based on partial cytochrome b and 16S mitochondrial DNA sequences. Copeia 2: 420–431.
López, A., W. Chen & W. Ortí, 2004. Esociform phylogeny. Copeia 3: 449–464.
Mace, G. M., H. P. Possingham & N. Leader-Williams, 2007. Prioritizing choices in conservation. In Macdonald, D. W. & K. Service (eds), Key Topics in Conservation Biology. Blackwell Publishing, Oxford: 17–34.
Machordom, A. & I. Doadrio, 2001. Evidence of a Cenozoic Betic–Kabilian connection based on freshwater fish phylogeography (Luciobarbus, Cyprinidae). Molecular Phylogenetics and Evolution 18: 252–263.
Marić, S., A. Snoj, N. Sekulić, J. Krpo-Ćetković, R. Šanda & V. Jojić, 2015. Genetic and morphological variability of the European mudminnow Umbra krameri (Teleostei, Umbridae) in Serbia and in Bosnia and Herzegovina—a basis for future conservation activities. Journal of Fish Biology 86(5): 1534–1548.
Marko, P. B. & M. W. Hart, 2012. Retrospective coalescent methods and the reconstruction of metapopulation histories in the sea. Evolutionary Ecology 26: 291–315.
Mangerud, J., M. Jakobsson, H. Alexanderson, V. Astakhov, G. K. C. Clarke, M. Henriksen, C. Hjort, G. Krinner, J. Lunkka, P. Möller, A. Murray, O. Nikolskaya, M. Saarnisto & J. Svendsen, 2004. Ice-dammed lakes and rerouting of the drainage of northern Eurasia during the Last Glaciation. Quaternary Science Reviews 23: 1313–1332.
Mikschi, E. & J. Wanzenböck, 1995. Proceedings of the First International Workshop on Umbra krameri. Annalen des Naturhistorischen Museums in Wien 97B: 438–508.
Miller, M. A., W. Pfeiffer & T. Schwartz, 2010. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the Gateway Computing Environments Workshop (GCE), 14 Nov. 2010. New Orleans: 1–8.
Moritz, C., K. McGuigan & L. Bernatchez, 2002. Conservation of freshwater fishes: integrating evolution and genetics with ecology. In Collares-Pereira, M. J., I. G. Cowx & M. M. Coelho (eds), Conservation of Freshwater Fishes: Options for the Future. Fishing News Books, Blackwell Science, Oxford: 293–310.
Nelson, G. J., 1972. Cephalic sensory canals, pitlines, and the classification of esocoid fishes, with notes on galaxiids and other teleosts. American Museum Novitates 2492: 1–49.
Olden, J. D., 2016. Challenges and opportunities for fish conservation in dam-impacted waters. In Closs, G. P., M. Krkosek & J. D. Olden (eds), Conservation of Freshwater Fishes. Cambridge University Press, Cambridge: 107–142.
Pekárik, L., J. Hajdú & J. Koščo, 2014. Identifying the key habitat characteristics of threatened European mudminnow (Umbra krameri, Walbaum 1792). Fundamental and Applied Limnology 184: 151–159.
Penck, A. & E. Brückner, 1909. Die Alpen im Eiszeitalter. Taunitz, Leipzig.
Pickens, D. C., 2003. Genetic Evidence for a Population Bottleneck in the Olympic Mudminnow (Novumbra hubbsi). B.S. Degree, University of Puget Sound, Tacoma.
Pritchard, J. K., M. Stephens & P. Donnelly, 2000. Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Rambaut, A., M. A. Suchard, D. Xie & A. J. Drummond, 2014. Tracer v1.6, Available from http://beast.bio.ed.ac.uk/Tracer. Accessed 1 Dec 2015.
Raykov, V. St., M. Panayotova, P. Ivanova, I. Dobrovolov & V. Maximov, 2012. First record and allozyme data of European mudminnow Umbra krameri Walbaum, 1792 (Pisces: Umbridae) in the Black Sea. Comptes rendus de l’Académie bulgare des Sciences 65(1): 49–52.
Sambrook, J., E. F. Fritseh & T. Maniatis, 1989. Molecular Cloning. A Laboratory Manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
Scotese, C. R., 2001. Atlas of Earth History, Volume 1, Paleogeography, PALEOMAP Project, Arlington.
Sekulić, N., S. Marić, L. Galamboš, D. Radošević & J. Krpo-Ćetković, 2013. New distribution data and population structure of the European mudminnow Umbra krameri in Serbia and Bosnia and Herzegovina. Journal of Fish Biology 83: 659–666.
Shedko, S. V., I. L. Miroshnichenko & G. A. Nemkova, 2013. Phylogeny of Salmonids (Salmoniformes: Salmonidae) and its molecular dating: analysis of mtDNA data. Russian Journal of Genetics 49: 623–637.
Skog, A., L. A. Vøllestad, N. C. Stenseth, A. Kasumyan & K. S. Jakobsen, 2014. Circumpolar phylogeography of the northern pike (Esox lucius) and its relationship to the Amur pike (E. reichertii). Frontiers in Zoology 11: 67.
Stepien C. A., J. Behrmann-Godel & L. Bernatchez, 2015. Evolutionary relationships, population genetics, and ecological and genomic adaptations of perch (Perca). In Couture, P., G. Pyle (eds), Biology of perch. CRC Press, Taylor and Francis Group, Boka Raton: 7–46.
Szpiech, Z. A., M. Jakobsson & N. A. Rosenberg, 2008. ADZE: a rarefaction approach for counting alleles private to combinations of populations. Bioinformatics 24: 2498–2504.
Takács, P., T. Erős, A. Specziár, P. Sály, Z. Vitál, Á. Ferincz, T. Molnár, Z. Szabolcsi, P. Bíró & E. Csoma, 2015. Population genetic patterns of threatened European Mudminnow (Umbra krameri Walbaum, 1792) in a fragmented landscape: implications for conservation management. PLoS ONE 10(9): e0138640.
Tamura, K. & M. Nei, 1993. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Molecular Biology and Evolution 10: 512–526.
Tamura, K., D. Peterson, N. Peterson, G. Stecher, M. Nei & S. Kumar, 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular Biology and Evolution 28: 2731–2739.
Thompson, J. D., T. J. Gibson, F. Plewniak, F. Jeanmougin & D. G. Higgins, 1997. The CLUSTAL_X windows interface, flexible strategies for multiple sequence alignment aided by quality analysis tool. Nucleic Acids Research 25: 4876–4882.
Tiffney, B. H., 1985. The Eocene North Atlantic Land Bridge: its importance in Tertiary and modern phytogeography of the Northern Hemisphere. Journal of the Arnold Arboretum 66: 243–273.
Trombitsky, I., V. Lobcenco & A. Moshu, 2001. The European mudminnow, Umbra krameri, still populates the Dniester River in Moldova. Folia Zoologica 50: 159–160.
Vähä, J. P., J. Erkinaro, E. Niemelä & C. R. Primmer, 2007. Life-history and habitat features influence the within-river genetic structure of Atlantic salmon. Molecular Ecology 16: 2638–2654.
Van Oosterhout, C., W. F. D. Hutchinson, P. M. Wills & P. Shipley, 2004. MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Molecular Ecology Notes 4: 535–538.
Velkov, B., L. Pehlivanov & M. Vassilev, 2004. Umbra krameri (Pisces: Umbridae): a reinstated species for the Bulgarian ichthyofauna. Acta Zoologica Bulgarica 56: 233–235.
Wakeley, J., 2000. The effects of subdivision on the genetic divergence of populations and species. Evolution 54: 1092–1101.
Wanzenböck, J., 1995. Current knowledge on the European mudminnow Umbra krameri Walbaum, 1792. Annalen des Naturhistorischen Museums in Wien 97B: 439–449.
Wanzenböck, J., 2004. European mudminnow (Umbra krameri) in the Austrian floodplain of the River Danube–Conservation of an indicator species for endangered wetland ecosystems in Europe. In Akcakaya, H. R., M. A. Burgman, O. Kindvall, C. C. Wood, P. Sjögren-Gulve, J. S. Hatfield & M. A. McCarthy (eds), Species Conservation and Management. Oxford University Press, New York: 200–207.
Wilson, M. V. H., D. B. Brinkman & A. G. Neuman, 1992. Cretaceous Esocoidei (Teleostei): early radiation of the pikes in North American fresh waters. Journal of Paleontology 66: 839–846.
Winkler, K. A. & S. Weiss, 2009. Nine new tetranucleotide microsatellite DNA markers for the European mudminnow Umbra krameri. Conservation Genetics 10: 1155–1157.
Yang, Z., 1993. Maximum likelihood estimation of phylogeny from DNA sequences when substitution rates differ over sites. Molecular Biology and Evolution 10: 1396–1401.
Acknowledgements
This study received support from the Slovenian Research Agency, the Ministry of Education, Science and Technological Development of the Republic of Serbia (Grant No. 173045) and the SYNTHESYS Project (CZ-TAF-5090; http://www.synthesys.info/) financed by European Community Research Infrastructure Action under the FP7 Integrating Activities Programme. RŠ was supported by the Ministry of Culture of the Czech Republic (DKRVO 2016/15, National Museum, 00023272) and DS by the DIANET postdoctoral fellowship programme (FP1527385002). TE, PT and ASp would like to express their thanks for Eszter Csoma for the DNA extraction of the Hungarian samples. Many thanks to dr Ladislav Pekarik for collected samples of European mudminnow from the Slovak Republic.
Author information
Authors and Affiliations
Corresponding author
Additional information
Handling editor: Christian Sturmbauer
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Marić, S., Stanković, D., Wanzenböck, J. et al. Phylogeography and population genetics of the European mudminnow (Umbra krameri) with a time-calibrated phylogeny for the family Umbridae. Hydrobiologia 792, 151–168 (2017). https://doi.org/10.1007/s10750-016-3051-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10750-016-3051-9