
Abstract
Eighteen isolates of the red algae Chondrus crispus were collected from Northern Atlantic sites, together with C. ocellatus, C. yendoi and C. pinnulatus from the North Pacific. The nuclear rDNA internal transcribed spacer (ITS) was sequenced and compared, spanning both the ITS regions and the 5.8S rRNA gene. Percentage of nucleotide variation for C. crispus ranged from 0.3% to 4.0%. Phylogenetic analyses were performed using maximum parsimony (MP), neighbor-joining (NJ) and minimum evolution methods. They showed that two main clades existed within the C. crispus samples examined and that suggested C. crispus had a single Atlantic origin. The clustering however did not follow the geographic origin. We hypothesized that the current distribution of C. crispus populations might be a result of three main factors: temperature boundaries, paleoclimate and paleoceanography. ITS data exhibited abundant molecular information not only for phylogeographical investigation but also for systematics studies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Chondrus crispus Stackhouse belongs to the division Rhodophyta, class Florideophyceae, order Gigartinales and family Gigartinaceae (Dixon & Irvine, 1977), and was first separated from the wide-embracing genus Fucus by Stackhouse (Stackhouse, 1797), and it was the basis of a multi-million dollar phycocolloid industry (Levring et al., 1969; Michanek, 1983). Usually, C. crispus flourishes in the lower shore and is distributed mainly on Atlantic coasts of Europe, such as Germany (Franklin et al., 2001), France (Gall et al., 1990; Boyen et al., 1994; Donaldson et al., 2000), Spain (Gutierrez & Fernández, 1992), the Netherlands (Groen et al., 1997), and Northern America like Canada (Chopin et al., 1996, 1997; Donaldson et al., 1998) and USA (Carrington et al., 2001; Zertuche-González et al., 2001), as well as in the Northern Russia, Western Baltic and Iceland (Dixon & Irvine, 1977). These surveys indicate that C. crispus is a cold temperate species restricted to the Northern Atlantic, and its distribution may be correlated with seasonal global isotherms (van den Hoek, 1984). This paper attempts to explain the contemporary distribution patterns of C. crispus based on other factors, such as climatological changes and vicariant events (Rögl & Steininger, 1984), and paleobiogeography (Bakker et al., 1995b), which greatly avail the recognition of global biogeographic disjunctions in marine organisms.
Nuclear ribosomal DNA (rDNA) sequences have been extensively used over the past decades to investigate phylogeographic questions among taxa at different hierarchical ranks in algae. At or below the species level, however, rDNA coding regions reach their limit or resolution (Hillis & Moritz, 1990; Bakker et al., 1992). For this reason, the greater sequence divergences of the internal transcribed spacer (ITS) regions of the rDNA are of interest (Bakker et al., 1992). Located between the three regions within the nuclear rDNA cistron (5′-18S-ITS1-5.8S-ITS2-28S-3′), the ITS regions are part of the initial transcript but spliced out later during maturation of pre-RNA (Gerbi, 1995; Mattai et al., 1993; van Nues et al., 1994). These regions are virtually free to evolve at a faster rate, as reflected in the number of substitutions and well-documented length variation (Yokota et al., 1989; Gardes et al., 1991). These characteristics allow to the usage of ITS for phylogenetic and biogeographic studies (Bakker et al., 1992, 1995a). In this study, we present a comparison of ITS of C. crispus sampled from eighteen sites along the North Atlantic Ocean, and three out-group isolates of C. ocellatus, C. yendoi and C. pinnulatus from the Northern Pacific Ocean. Our aims were to describe phylogenetic relationships among distant isolates of C. crispus, to compare the ITS sequence divergence of morphologically different isolates from various sites, and to analyze the dispersion mechanisms of C. crispus populations in the view of Tethyan events.
Materials and methods
Sample collection
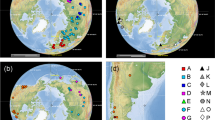
Eighteen samples of C. crispus Stackhouse were collected from seventeen sites in the Atlantic Ocean, ranging from Lepreau, Canada, situated at 45° N, 66° W, to Helgoland, situated at 54° N, 8° E in Germany (Fig. 1). Three Pacific species (C. ocellatus Holmes, C. yendoi Yamada & Mikami and C. pinnulatus (Harvey) Okamura) were also included. Voucher specimens were prepared and lodged in KEMB (Key Laboratory of Experimental Marine Biology, Institute of Oceanology, Chinese Academy of Sciences). A mid portion of each specimen was selected for DNA extraction and further analysis. Where isolates collected from the same locality exhibited similar morphology, only one individual was used for sequencing. The two morphological variants of C. crispus (marked with #1 and #2) from Roscoff, France were analyzed separately. All steps including DNA extraction, PCR and sequencing were repeated three times. Voucher images of representative isolates of each entity are presented in Figs. 2–14.

A map indicates the collection sites of Chondrus crispus in this study

(2) Prince Edward Island, Canada; (3) Helgoland North sea, Germany Eastney, Portsmouth, UK; (4) Eastney, Portsmouth, UK; (5) Hayling Island, UK; (6) New Brunswick; (7) Portaferry, Northern Ireland; (8) Portsall, France; (9) Roscoff, France (isolate #1); (10) Roscoff, France (isolate #2); (11) St. Vaast La Hougue, France; (12) Sune, Porto, Portugal; (13) Wembury beach, Devon, England; (14) Buarocos bay, Portugal. The scale bar shows 1 cm
DNA extraction
DNA was isolated according to the procedure described by Hu et al. (2004). In this procedure, about 0.12 g fresh tissue was frozen in liquid nitrogen, ground into fine powder and quickly transferred to a 1.5 ml tube containing 0.7 ml extraction buffer (0.1 M Tris–HCl, pH 8.0, 0.05 M ethylene diamine tetracetic acid (EDTA), 0.5 M NaCl, 1.6% sodium dodecyl sulphate [SDS], 0.2% polyvinyl-polypyroolidone [PVPP] and 2% β-mercaptoethanol) and incubated at 37°C for 1 h. An equal volume of ice-cold potassium acetate (5.0 M, pH 7.5) was added and mixed. The content were kept on ice for 20 min and was centrifuged at 10,600 × g for 15 min. The aqueous phase was collected and extracted with an equal volume of phenol:chloroform:isoamyl alcohol (25:24:1) and chloroform:isoamyl alcohol (24:1) for two times. The supernatant was collected and RNAse was added to a final concentration of 0.1 mg ml−1. The upper aqueous was extracted with chloroform:isoamyl alcohol (24:1) and 2/3 volume of ice-cold isopropanol was added to precipitate DNA. Finally, the DNA was washed with 70% EtOH and stored at −20°C.
ITS region amplification and sequencing
The nuclear ITS rDNA was amplified using the synthetic primers TW18: 5′GGGATCCGTTTCCGTAGGTGAACCTGC3′ (Annealing to 3′ end of 18S rDNA) and AB28: 5′GGGATCCATATGCTTAAGTTCAGCGGGT3′ (Annealing to the 5′ end of 28S rDNA) according to Goff & Moon (1993). Amplification was performed on an Eppendorf Masterthermocycler Personal (Germany). The reaction mixture (20 μl) contained 10 mM Tris · HCl (pH 8.3), 50 mM KCl, 1.5 mM MgCl2, 2.0 mM dATP, dCTP, dGTP, dTTP respectively, 50 ng nuclear DNA, 1.0 mM of each primer and 1.0 U Taq DNA polymerase (Takara Biotechnology Co., Ltd. Japan). The PCR parameters were 5 min at 95°C; 1 min at 90°C and 2 min at 50°C for 5 cycles; 72°C for 1 min; followed by 30 cycles of 1 min at 90°C, 1 min at 60°C, 1 min at 72°C; finally left at 72°C for 10 min. The size, purity and yields of PCR products were assessed by electrophoresis through a 1.5% agarose gel for 1 h at 80 V and stained with ethidium bromide. Reaction products were purified with QIAquick PCR purification kit (QIAGEN, Germany) and sequenced on an ABI Prism 377 automatic sequencer (Perkin ElmerApplied Biosystems, China) utilizing standard methods. Sequencing primers were the amplification primer AB28 and revised TW18-1: 5′CGTTTCCGTAGGTGAACC3′.
Sequence identification and alignment
The boundaries of ITS regions and the 18S, 5.8S, and 28S rDNA were determined by comparing aligned sequences of the ITS spacer region of Gracilariopsis and Gracilaria with GenBank accession no. U21339-U21345 (Goff et al., 1994), Mazzaella species with AY225230-AY225290 (Ross et al., 2003) and some other Gigartinales species with AF400051-AF400062, and AF398529-AF398547 (Hughey et al., 2001). Sequences were compared using the GCG software package (Genetics Computer Group, 1994) and aligned with Clustal X (Thompson et al., 1997). The final adjustments were conducted manually. In order to assess nucleotide substitution patterns, substitutions were counted in pair-wise comparison among all sequences using MEGA 3.0 (Kumar et al., 2004) regarding the gaps as missing data. Three other Chondrus species C. ocellatus, C. yendoi, C. pinnulatus from the Pacific Ocean, and M. splendens (Setchell et Gardner) Fredericq (Ross et al., 2003) with accession no. AY225290 were used as out-groups.
Phylogenetic analysis
For minimizing information loss, gaps and missing data were deleted only on a pair-wise basis and not down an entire column. Tamura–Nei model was used to estimate the genetic divergence with software MEGA 3.0. Pair-wise distance, Tajima’s Neutrality Test (Tajima, 1989) and Interior branch test using minimum evolution (ME) (Rzhetsky & Nei, 1993) and neighbor-joining (NJ) methods were calculated with 1,000 replications, respectively. Phylogenies were inferred with maximum parsimony (MP) and NJ (Kimura-2 model) methods, gaps/missing data were treated as complete deletions. For maximum likelihood (ML) analysis, 1,000 replicate data sets generated by SEQBOOT from PHYLIP 3.57c (Felsenstein, 1995) were subject to distance analyses, the DNAPARS and DNAML program was used to analyze the 1,000 replicate data sets by parsimony (jumble = 5). Bootstrapping was also performed with 1,000 re-samplings for MP analyses in PHYLIP. The three phylogenetic trees were drawn by Tree View program. FOLD programs from the UWGCG package and visual inspection were used to investigate the presence of secondary structures in the ITS sequences (Bakker et al., 1992).
Results
Sequence data
GenBank accession numbers for the ITS sequences of each isolate identified in the study, along with the collection information are listed in Table 1. Alignment of complete variable nucleotides of ITS1 and ITS2 including the 5.8S rRNA gene were obtained for all isolates (Fig. 15). ITS length ranged between 148 and 151 base pairs for ITS1, and between 399 and 404 base pairs for ITS2. Average nucleotide composition of all isolates was approximately 34.3% and 44.5% GC in ITS1 and ITS2. The total ITS length was about 710 base pairs. The final in-group alignment including C. ocellatus, C. yendoi and C. nipponicus contained 745 positions, of which 101 are variable, among them 39 are phylogenetically informative, and the maximum sequence divergence is 13.56% and 5.24%, respectively.

Alignment of ITS region of seventeen C. crispus isolates and three outgroups, numbers indicated variant nucleotides in different sites and the identical nucleotides were not shown
Transitions were about equal to transversions, and the percentage of nucleotide variation for C. crispus ranged from 0.3% to 4.0%. Table 2 shown that pair-wise distances varied from 0.0% to 3.6%, the minimum distance was among the four isolates from France (PS, DE, RS #1 and #2), and the maximum value was among the two isolates from Canada (NB) and France (SVLH), respectively.
Phylogenetic analysis
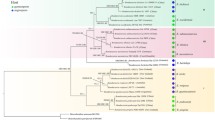
The three phylogenetic reconstruction methods (NJ, MP and ML) produced similar tree topologies. Maximum parsimony produced 82 equally most parsimonious trees of 517 steps. For ML analysis, the empirical base frequencies were unequal (A = 0.27621, C = 0.18420, G = 0.25328, T = 0.28361), −Ln Likelihood = 2956.7969. In three phylogenetic dendrograms, there were two major clades within the C. crispus group (Figs. 16, 17). The first clade contained two isolates (SP, BB) from Portugal, PF from Northern Ireland, DP from Wales, NB from Canada, three isolates (SVLH, RS #2 and FB) from France and three isolates (WBD, BR and HI) from England, respectively with moderate bootstrap support (55%); while the three isolates (RS #1, DE and PS) from France, PEI from Canada, EP from UK, I from Ireland and HNS from Germany formed the second clade with moderate support (58%). In clade one, WBD and HI had closest relationships (89%), and in group two, three isolates (EP, HNS and RS #1) had sister relationships with high bootstrap support (96%). Outgroups, C. ocellatus, C. pinnulatus and C. yendoi from North Pacific Ocean were clearly distinguished from C. crispus.

The NJ tree generated from ITS sequences of C. crispus using interior branch test. Numbers above the branches are bootstrap values of 1,000 replicates, C. pinnulatus, C. yendoi, C. ocellatus and M. splendens were used as outgroups

The consensus tree inferred from interior branch test with ME method (1,000 replications)
Discussion
As shown in Figs. 2–14, it was interesting that C. crispus exhibited conspicuous morphological variability which was not reflected by the ITS sequence divergence or pair-wise distance (Table 2), although base mutation including transition/transversion and insertion/deletion might be responsible for the variability. More likely is that the diversity of morphology of C. crispus was due to external environmental parameters like tidal variation, exposure to water current etc. (Gutierrez & Fernandez, 1992). For an instance, at low intertidal and exposed sites, C. crispus is characterized by fewer dichotomies and exhibits filiform morphology, while at high intertidal sites in sheltered areas, C. crispus is characterized by more dichotomies, smaller sizes and narrow fronds (Gutierrez & Fernandez, 1992). It seems likely that the morphological variation seen in C. crispus mainly from external environmental factors.
ITS data and phylogeny
The ITS regions were chosen for analysis because they provided a measure of DNA variation that was appropriate for species or subspecies levels of comparisons (Gardes et al., 1991; Lee & Taylor, 1992). In plant genomes, nuclear ribosomal genes are organized in clusters of tandemly repeated units, and thousands of ITS copies are present and located in one or several chromosomes. Concerted evolution of intra- and interchromosomal loci, a process that takes 1–2 million years, is believed to result in homogenization of individual repeats (Won & Renner, 2005). So, it plays an important role in phylogenetic applications (Zimmer et al., 1989; Hillis & Moritz, 1990; Li & Graur, 1991).
Chopin et al. (1996) examined seven different isolates of C. crispus from different locations across both the Maritime Province, Canada and Europe by sequencing the ITS region. They found 2.18% sequence divergence for the two isolates from Nova Scotia and PEI, which exceeded by far the average intraspecific sequence divergence (1.3%) of ITS1 region for five various red algal genera (Goff et al., 1994, 1996). They suggested that the genetic diversity of C. crispus neither correlated with the phenotype for the plants investigated nor the geographic origin. However, in this research we found that the intraspecific sequence in C. crispus varied from 0.3% to 4.0%, this is adequate for the study of phylogeography. It was interesting that there were relatively low pair-wise distances in C. crispus species (0–0.16%) except the isolate (SVLH) from France (Table 2), which showed higher distances (0.23–0.36%) to all other isolates. The ITS sequence may not be correlated with the geographic distance of the isolates, and further studies are needed to explain the divergence.
It is interesting that, in some ITS regions of C. crispus, short repetitive motifs were found. The presence of these motifs is most likely due to the process of slip-strand mispairing events, causing the addition or deletion of repeat units during replication (Levinson & Gutman, 1987), i.e. an intrahelical event rather than an interhelical event and displacement of the strands of a DNA duplex followed by mispairing at the site of an existing short tandem repeat. Over time, this leads to insertions or deletions of one or several short repeats that can form distinctive nested patterns. The process can be self-accelerating and maintained for long periods in specific populations. It has also been hypothesized that this might provide the raw material for the evolution of regulatory elements, especially when located near genes (Levinson & Gutman, 1987; Bakker et al., 1992).
One factor that may affect ITS evolution is secondary structure. Bakker et al. (1995b) studied the Cladophora albida/sericea complex and found the conservative regions within both ITS1 and ITS2 correlated with double-stranded regions in the ITS secondary structure (within the pre-rRNA molecule). Van Nues et al. (1994) demonstrated that distinct conserved structures existed within ITS1 directly processing of pre-rRNA (precursor RNA). No such clear secondary structural elements, however, could be found in the C. crispus ITS sequences. Another factor is the variability of the ITS sequence, which has been proved to strongly correlate with sexual isolation in the Volvocacean algae Pandorinum morum Bory and Gonium pectorale Mueller (Coleman et al., 1994). Geographically disparate isolates of G. pectorale can interbreed and this species has an ITS region that is remarkably uniform with a world-wide distribution. Isolates of P. morum behave quite differently, and syngens (breeding group) of worldwide distribution or those restricted to one small geographical area show great similarity for ITS sequences. Since little is known about reproductive patterns or sexual isolation in C. crispus, we cannot decide whether a similar explanation applies to the ITS evolution in this species.
Biogeography
The topology and level of divergence expressed in the trees (Figs. 16, 17) is consistent that of a relative old species in which C. crispus from Northern Atlantic are of more recent origin. The most general biogeographic conclusion that can be drawn from the consistently more basal position of three Chondrus speices from the Pacific, is of a Pacific origin. However, while a general theory explaining the pattern of biogeographic distribution for algae along the Northern Atlantic coasts is presently lacking, we can implicate with confidence the roles of paleoclimate and paleoceanography as prime factors influencing the biogeographic distribution of C. crispus. Neumayr (1885) hypothesized a primordial “Central Mediterranean”, which arose ca. 160 million years ago and extended from Mexico to the Himalayas. Suess (1893) renamed this great equatorial ocean the Tethys Sea, which provided an open east–west connection between the present-day Atlantic and Pacific Oceans via the “Mediterranean” until roughly 12 Ma ago (Tarling, 1980; Rögl & Steininger, 1984; Por, 1989). Also, biogeographic disjunctions observed today may be explained by Tethyan closure events (Silva, 1962; Rögl & Steininger, 1984). So, the biogeographical consequences of plate movements, interactions, the rearrangement of continental landmasses and island areas, the opening and closing of sea and ocean basins initiated by these movements and interactions have profoundly affected the distribution of organisms on land and in the ocean. In this study, we assumed that both in clade one and two, the isolates from Canada grouped with the isolates from France, and United Kingdom. This affinity showed that the isolates from Canada, located in the western Atlantic were closely related to the isolates from France, England, Portugal and Germany, located in eastern Atlantic. This is congruent with the Tethyan hypothesis that an early equatorial cosmopolitan marine flora gradually became dispersed.
From this point onward in the discussion, we can therefore assume that an important paleoceanographic event was the closure of the warm Tethys Sea at around 12 Ma ago, thus “pinching off” the major Pacific and Atlantic domains, and the subsequent further division of the eastern and western Atlantic marine floras (van den Hoek, 1984; Joosten & van den Hoek, 1986). Tethyan divergence has been suggested for many green algae in which large genetic distances have been found, such as Dictyosphaeria cavernosa Boergesen (Olsen et al., 1987), Cladophora and related species (Kooistra et al., 1992; Bakker et al., 1995a, b). Given the large uncertainties about the divergence times, the estimates are useful as a qualitative pre- or post-Tethys closure divergence estimate at most. At the same time, we can understand the distribution of the algae and the formation of the algal regions and provinces from a historical point of view (van den Hoek, 1975). The Atlantic Ocean opened up in the Cretaceous (135–65 Ma ago), so that a distinct Caribbean province could originate in the tropical Tethyan Realm. To the south and to the north of this tropical Tethyan Realm, (a belt between the 50° S to 50° N), conditions were warm and temperate. During the Cretaceous polar temperatures fluctuated between 10°C and 16°C. By the early tertiary polar temperatures may have dropped to the point where the development of cold temperature flora could have begun (with winter surface temperature at Spitsbergen of 5–8°C) (van den Hoek, 1975). From this period onwards, there were a number of climatic temperature fluctuations. A gradual cooling culminated in the Pleistocene ice ages (2–3 Ma ago), this affected the distribution of marine floras.
Tropical taxa were largely unaffected, while temperate floras were forced south or became extinct depending on their eurythermic abilities. Retreat of the ice sheets meant a mass recolonization of the middle and North Atlantic (and Mediterranean) of taxa whose distribution patterns follow isotherms rather than latitude (MacIntyre, 1981). This may then explain the present complex dispersion of C. crispus in Atlantic Ocean. About 135–65 Ma ago, North America and Europe were moving apart slowly. After that, the warm Tethys Sea was closed at around 12 Ma ago, thus “pinching off” the major Pacific and Atlantic domains, and the subsequent further division of the eastern and western Atlantic marine floras. The British Isles was also separated from European continent, then C. crispus formerly distributed in Northern Pangaea dispersed to both east and west of the Atlantic Ocean.
Further speculations might include the effects of vicariance, temperature boundaries and dispersal. The correlation between thermal barriers and vicariance events seen indicate that the former also has play a role because each would have predicted a correlation between biogeographic distribution and tree topology (Bakker et al., 1995a). Joosten & van den Hoek (1986) divided the distribution of 613 red algal genera into 22 climatically defined coastal regions two of which were the cold temperate Northern (CN3 and CN4) and the warm temperate Western (WN3 and WN4) Atlantic region (70–30° N). C. crispus has a range similar to this (northern Russia (65° N) to southern Spain (38° N)) (Dixon & Irvine, 1977). We think the most environmental factor responsible for the distribution over these areas is temperature. In New Hampshire, USA, C. crispus grows abundantly in waters with an annual variation in surface temperature from −1°C to 19°C (Mathieson & Burns, 1975), and its optimum temperature for growth has been reported as 10–15°C (Fortes & Luning, 1980), 15–17°C (Tasende & Fraga, 1999) and 20°C (Simpson & Shacklock, 1979). Prince & Kingsbury (1973) reported cessation of growth in C. crispus culture at 26°C, first mortality of spores at 21.1°C and total mortality of spores at 35°C, even if exposed for just 1 min. Maximum sea surface temperature in the Northern Atlantic rarely exceeds 20°C (Joosten & van den Hoek, 1986). It is unlikely that C. crispus will colonize the middle tropical East and West Atlantic regions, where the summer temperature is 25°C. C. crispus is not capable of completing all the stages of its life cycle at these temperatures.
In conclusion, ITS sequence data suggests a single phylogenetic Atlantic origin for C. cripus (Atlantic origin). The present distribution is attributable to temperature barriers, vicariance, paleoceanography and paleoclimate. The results indicate that C. crispus distributed from cold to warm-temperate areas, might be useful in understanding historical vicariance in marine algae, and it may provide further illumination on continent drift.
References
Bakker, F. T., J. L. Olsen, W. T. Stam & C. van den Hoek, 1992. Nuclear ribosomal DNA internal transcribed spacer regions (ITS1 and ITS2) define discrete biogeographic groups in Cladophora albida (Chlorophyta). Journal of Phycology 28: 839–845.
Bakker, F. T., J. L. Olsen & W. T. Stam, 1995a. Global phylogeography in the cosmopolitan species Cladophora vagabunda (Chlorophyta) based on nuclear rDNA internal transcribed spacer sequences. European Journal Phycology 30: 197–208.
Bakker, F. T., J. L. Olsen & W. T. Stam, 1995b. Evolution of nuclear rDNA ITS sequences in the Cladophora albida/sericea clade (Chlorophyta). Journal of Molecular Evolution 40(6): 640–651.
Boyen, C., C. Leblane, B. Kloareg & S. Loiseaux-de Goer, 1994. Physical map and gene organization of the mitochondrial genome of Chondrus crispus (Gigartinales, Rhodophyta). Plant Molecular Biology 26: 691–697.
Carrington, E., S. P. Grace & T. Chopin, 2001. Life history phases and the biochemical properties of the red algae Chondrus crispus (Rhodophyta). Journal of Phycology 37: 699–704.
Chopin, T., C. J. Bird, C. A. Murphy, J. A. Osborne, M. U. Patwary & J. Y. Floc’h, 1996. A molecular investigation of polymorphism in the North Atlantic red alga Chondrus crispus (Gigartinales). Phycol. Res. 44: 69–80.
Chopin, T., H. Lehmal & K. Halcrow, 1997. Polyphosphates in the red macroalgae Chondrus crispus (Rhodophyceae). New Phytologist 135: 587–594.
Coleman, A. W., A. Suarez & L. J. Goff, 1994. Molecular delineation of species and syngens in Volvocacean green algae (Chlorophyta). Journal of Phycology 30: 80–90.
Dixon, P. S. & L. M. Irvine, 1977. Seaweeds of the British Isles, Vol. 1. Rhodophyta, Part 1: Introduction, Nemaliales, Gigartinales. British Museum (Natural History), London.
Donaldson, S. L., T. Chopin & G. W. Saunders, 1998. Amplification fragment length polymorphism (AFLP) as a source of genetic markers for red algae. Journal of Applied Phycology 10: 365–370.
Donaldson, S. L., T. Chopin & G. W. Saunders, 2000. An assessment of the AFLP method for investigating population structure in the red alga Chondrus crispus Stackhouse (Gigartinales, Florideophyceae). Journal of Applied Phycology 12: 25–35.
Felsenstein, J., 1995. PHYLIP (Phylogeny Inference Package), Version 3.57c, Distributed by the author. Department of Genetics, University of Washington, Seattle, USA.
Fortes, M. D. & K. Luning, 1980. Growth rates of North Sea macroalgae in relation to temperature, irradiance and photoperiod. Helgolander Meeresuntersuchungen 34: 15–29.
Franklin, L. A., G. Krabs & R. Kuhlenkamp, 2001. Blue light and UV-A radiation control the synthesis of mycosporine-like amino acids in Chondrus crispus (Florideophyceae). Journal of Phycology 37: 257–270.
Gall, Y. L., J. P. Braud & B. Kloareg, 1990. Protoplast production in Chondrus crispus gametophytes (Gigartinales, Rhodophyta). Plant Cell Reports 8: 582–585.
Gerbi S. A., 1995. Small nucleolar RNA. Biochemistry and Cell Biology 73: 845–858.
Gardes, M., T. J. White, J. A. Fortin, T. D. Bruns & J. W. Taylor, 1991. Identification of indigenous and introduced symbiotic fungi in ectomycorrhizae by amplification of nuclear and mitochondrial ribosomal DNA. Canadian Journal of Botany 69: 180–190.
Goff, L. J. & D. A. Moon, 1993. PCR amplification of nuclear and plastid genes from algal herbarium species and algal spores. Journal of Phycology 29: 381–384.
Goff, L. J., D. A. Moon & A. W. Coleman, 1994. Molecular delineation of species and species relationships in the red algal agarophytes Gracilariopsis and Gracilaria (Gracilariales). Journal of Phycology 30: 521–537.
Goff, L. J., D. A. Moon, P. Nyvall, B. Stache, K. Mangin & G. Zuccarelo, 1996. The evolution of parasitism in the red algae: molecular comparsions of adelphoparasites and their hosts. Journal of Phycology 32: 297–312.
Groen, B. W., S. D. Vries & J. A. Duine, 1997. Characterization of hexose oxidase from the red seaweed Chondrus crispus. European Journal of Biochemistry 244: 858–861.
Gutierrez, L. M. & C. Fernandez, 1992. Water motion and morphology in Chondrus crispus (Rhodophyta). Journal of Phycology 28: 156–162.
Hillis, D. M. & G. Moritz, 1990. Molecular Systematics. Sinauer Assoc., Sunderland, Massachusetts.
Hu, Z. M., X. Q. Zeng, A. H. Wang, C. J. Shi & D. L. Duan, 2004. An efficient method for DNA isolation from red algae. Journal of Applied Phycology 16: 161–166.
Hughey, J. R., P. C. Silva & M. H. Hommersand, 2001. Solving taxonomic and nomenclatural problems in Pacific Gigartinaceae (Rhodophyta) using DNA from type material. Journal of Phycology 37: 1091–1109.
Joosten, A. M. T. & C. van den Hoek, 1986. World-wide relationships between red seaweed floras: a multivariate approach. Botanica Marina 29: 195–214.
Kooistra, W. H. C. F., W. T. Stam, J. L. Olsen & C. van den Hoek, 1992. Biogeography of the green algae Cladophoropsis membranacea (Chlorophyta) based on nuclear rDNA ITS sequences. Journal of Phycology 28: 660–668.
Kumar, S., K. Tamura & M. Nei, 2004. MEGA3: Integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Briefings in Bioinformatics 5: 150–163.
Lee, S. B. & J. W. Taylor, 1992. Phylogeny of five fungus-like protoctistan Phytophthora species, inferred from the internal transcribed spacers of ribosomal DNA. Molecular Biology and Evolution 9: 636–653.
Levison, G. & G. A. Gutman, 1987. Slipped-strand mispairing: a major mechanism for DNA sequence evolution. Molecular Biology and Evolution 4: 203–221.
Levring, T., H. A. Hoppe & O. J. Schmid, 1969. Marine algae, a survey of research and utilization. Cram de Gruyter and Company, Hamburg.
Li, W. H. & D. Graur, 1991. Fundamentals of molecular evolution. Sinauer Assoc., Sunderland, Massachusetts.
MacIntyre, A., 1981. Seasonal reconstructions of the earth’s surface at the last glacial maximum. Geol. Soc. Am. Map and Chart Series, MC-36.
Mathieson, A. C. & R. L. Burns, 1975. Ecological studies of economic red algae. V. Growth and reproduction of natural and harvested populations of Chondrus crispus Stackhouse in New Hampshire. Journal of Experimental Marine Biology and Ecology 17: 137–156.
Mattai, I. W., D. Tollervey & B. Seraphin, 1993. Small nuclear RNAs in messenger RNA and ribosomal RNA processing. FASEB Journal 7: 47–53.
Michanek, G., 1983. World resources of marine plants. Benthic Plants: In Marine Ecology, 5(2): 795–837. Chichester, UK.
Neumayr M., 1885. Die geographische Verbreitung der Juraformation. Denks. Akad. Wissensch. Math. Nat. Cl. 15: 57–144.
Olsen, J. L., W. T. Stam, P. V. M. Bot & C. van den Hoek, 1987. Single copy DNA-DNA hybridization studies in Pacific and Caribbean isolates of Dictyosphaeria cavernosa (Chlorophyta) indicate a long divergence. Helgolander Wissenschaftliche Meeresuntersuchungen 41: 377–383.
Por, F. D., 1989. The legacy of Tethys: Aquatic Biogeography of the Levant. Kluwer Acad. Publ., Dordrecht, The Netherlands.
Prince, J. S. & J. M. Kingsbury, 1973. The ecology of Chondrus crispus at Plymouth, Massachusetts. III. Effect of elevated temperature on growth and survival. Biological Bulletin 145: 580–588.
Rögl, F. & F. F. Steininger, 1984. Neogene paratethys, Mediterranean and Indo-Pacific Seaways. Implications for the paleobiogeography of marine and terrestrial biotas. Fossil and Climate (Bzenchley, P.J., Editor) Chichester, 171–200.
Ross, P. J., S. L. Donaldson & G. W. Saunders, 2003. A molecular investigation of Mazzaella (Gigartinales, Rhodophyta) morphologically intermediate between Mazzaella linearis and M. splendens. Botanica Marina 46: 202–213.
Rzhetsky, A. & M. Nei, 1993. Theoretical foundation of the Minimum-Evolution method of phylogenetic inference. Molecular Biology and Evolution 10(5): 1073–1095.
Silva P. C., 1962. Comparison of algal florisitic patterns in the Pacific and those in the Atlantic and Indian Oceans, with special reference to Codium. Proc. IX Pac. Sci. Congr. 4: 201–216.
Simpson, F. J. & P. F. Shacklock, 1979. The cultivation of Chondrus crispus. Effect of temperature on growth and carrageenan production. Botanica Marina 22: 295–298.
Stackhouse, J., 1797. Nereis Brittanica, 31–70. Fascicle 2, Bathoniae and Londini.
Suess, E., 1893. Are great ocean depths permanent? Natural Science 2: 180–187.
Tajima F., 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595.
Tarling, D. H., 1980. Continental drift and biological evolution. Carol. Biol. Readers 113: 1–31.
Tasende, M. G. & M. I. Fraga, 1999. The growth of Chondrus crispus Stackhouse (Rhodophyta, Gigartinaceae) in Laboratory culture. Ophelia 51: 203–213.
Thompson, J. D., T. J. Gibson, F. Plewniak, F. Jeanmougin & D. G. Higgins, 1997. The Clustal X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Research 24: 4876–4882.
Van, den Hoek C., 1975. Phytogeographic provinces along the coasts of the Northern Atlantic Ocean. Phycologia 14(4): 317–330.
Van, den Hoek C., 1984. World wide latitudinal and longitudinal seaweed distribution patterns and their possible causes, as illustrated by the distribution of Rhodophytan genera. Helgolander Wissenschaftliche Meeresuntersuchungen 38: 227–257.
Van, Nues R. W., J. M. J. Rientjes, C. A. F. M. Van Der Sande, S. F. Zerp, C. Sluiter, J. Venema, R. J. Plania & H. A. Raus, 1994. Separate structural elements within internal transcribed spacer 1 of Saccharomyces cerevisiae precursor ribosomal RNA direct the formation of 17S and 16S rRNA. Nucleic Acids Research 22: 912–919.
Won, H. & S. S. Renner, 2005. The internal transcribed spacer of nuclear ribosomal DNA in the gymnosperm Gnetum. Molecular Phylogenetics and Evolution 36: 581–597.
Yokota, Y., T. Kawata, Y. Iida, A. Kato & S. Tanifuji, 1989. Nucleotide sequences of the 5.8S rRNA gene and internal transcribed spacer regions in carrot and broad bean ribosomal DNA. Journal of Molecular Evolution 29: 294–301.
Zertuche-Gonzalez, J. A., G. Garcia-Lepe, I. Pacheco-Ruiz, A. Chee, V. Gendrop & J. M. Guzman, (2001). Open water Chondrus crispus Stackhouse cultivation. Journal Applied Phycology 13: 249–253.
Zimmer, E. A., R. K. Hamby, M. L. Arnold, C. A. Leblanc & E. C. Theriot, 1989. The Hierarchy of life. Ribosomal RNA phylogenies and flowering plant evolution (Fernholm, B., Bremer, K., Jornvall, H., Editors), 70: 205–214. Elsevier, Amsterdam, The Netherlands.
Acknowledgements
We thank Dr. L. A. Franklin (Germany), Dr. G. W. Saunders (Canada), Dr. M. Dring (Northern Ireland), Dr. M. Brown (England), Dr. L. Pereira (Portugal), Dr. S. Costa (Portugal), Dr. F. Weinberger (France), Dr. S. Conan (France), Dr. W. F. Farnham (UK), Dr. T. Motomura (Japan) for their help in collecting specimens. This research was supported by Shandong Agriculture Seedstocks Project, Knowledge Inovation Program of the Chinese Academy of Sciences, and the Key Laboratory of Experimental Marine Biology, IOCAS.
Author information
Authors and Affiliations
Corresponding author
Additional information
Handling editor: C. Sturmbauer
Rights and permissions
About this article
Cite this article
Hu, Z., Zeng, X., Critchley, A.T. et al. Phylogeography of the Northern Atlantic species Chondrus crispus (Gigartinales, Rhodophyta) inferred from nuclear rDNA internal transcribed spacer sequences. Hydrobiologia 575, 315–327 (2007). https://doi.org/10.1007/s10750-006-0381-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10750-006-0381-z