
Abstract
Cardiac dysfunction is a well-recognized complication of severe sepsis and septic shock. Cardiac dysfunction in sepsis is characterized by ventricular dilatation, reduction in ejection fraction and reduced contractility. Initially, cardiac dysfunction was considered to occur only during the “hypodynamic” phase of shock. But we now know that it occurs very early in sepsis even during the “hyperdynamic” phase of septic shock. Circulating blood-borne factors were suspected to be involved in the evolution of sepsis induced cardiomyopathy, but it is not until recently that the cellular and molecular events are being targeted by researchers in a quest to understand this enigmatic process. Septic cardiomyopathy has been the subject of investigation for nearly half a century now and yet controversies exist in understanding it’s pathophysiology. Here, we discuss our understanding of the pathogenesis of septic cardiomyopathy and the complex roles played by nitric oxide, mitochondrial dysfunction, complements and cytokines.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Myocardial depression is a major contributor to mortality and morbidity in patients with sepsis. Contractile dysfunction in the septic heart is manifest as biventricular dilatation, reversible decrease in ejection fraction, diminished blood pressure response to IV fluids, and blunted ability to augment cardiac output despite increased levels of circulating catecholamines [1]. Traditionally, the physiologic disturbances have classically been described in a biphasic spectrum: early hyperdynamic shock characterized by increased cardiac output, decreased systemic vascular resistance (SVR) and warm, perfused skin, followed by cold hypodynamic shock, during which SVR increases to compensate for worsened cardiac output, resulting in tissue hypoperfusion, cool skin and eventual organ failure [1]. However, recent research suggests that hypodynamic shock is a mere manifestation of inadequate volume resuscitation and may be prevented by appropriate volume loading [2].
Experimental evidence indicates that even patients with so-called hyperdynamic shock exhibit myocardial dysfunction relative to non-septic controls [3]. These patients have lower stroke work indices (stroke volume x mean arterial blood pressure, standardized for body surface area) as a function of end-diastolic volume, marking a downward and rightward shift of the Frank-Starling mechanism [4]. In fact, dilatation of the left ventricle in this setting is seen as an adaptive response that preserves cardiac output via the Starling mechanism, and it has been associated with lower mortality and improved prognosis in patients with sepsis [5]. Earlier studies [6] note survivors of septic cardiomyopathy have significantly more dilated left ventricles (LV) and decreased ejection fractions (EF) relative to non-survivors [6]. However, newer literature has shown that lower ejection fractions translate into poorer prognoses [2].
In recent years, investigators have focused their attention on cytokines as the possible mediators of the myocardial depression of sepsis. Several types of nucleated cells produce and locally release these proteins in response to surgical, traumatic, ischemic or septic insults [7]. Studies performed on rodent models of sepsis show significant decreases in measures of contractility in cardiomyocytes exposed in vitro to TNF-α, IL-1β [8], and IL-6 [9]. Accordingly, patients with sepsis demonstrate higher levels of interleukins and complement components in their bloodstream [2].
The cellular mechanisms underlying cytokine-mediated cardiomyopathy are not entirely clear. Inducible nitric oxide (NO) production, intracellular calcium currents, oxidation–reduction imbalance, and disrupted respiratory chain activities have all been invoked in explanation. The remainder of this review will focus on the physiology of septic cardiomyopathy. We will review normal excitation–contraction (E-C) coupling in the cardiomyocyte in order to illustrate possible mechanisms of disturbance. Special attention will be directed toward the interplay between endothelial NO expression and sarcoplasmic calcium currents, as well as the roles of cytokines.
Normal E-C coupling
Myocardial contraction and relaxation is regulated by the interplay among contractile proteins (thin actin and thick myosin filaments), regulatory proteins (troponin and tropomyosin) and calcium ions. In the resting state, tropomyosin blocks the site of actin–myosin interaction, while the troponin-I subunit inhibits the actin–myosin ATPase (Fig. 2) [10]. Extracellular calcium (Ca) enters the myocyte through sarcolemmal L-type Ca channels (LTCC) during phase 2 of the cardiac action potential. This, in turn, triggers stored intracellular Ca release from the sarcoplasmic reticulum (SR) into the cytoplasm via ryanodine receptors (RyR) (Fig. 1). As the intracellular Ca concentration increases, the ion binds the c-subunit of troponin, leading to conformational changes that relieve troponin and tropomyosin inhibition. Actin and myosin are then free to crosslink and contraction proceeds with ATP hydrolysis (Fig. 2) [10]

Role of calcium in normal cardiomyocyte contraction
.

Actin–myosin interaction at rest and during contraction
Ca channel closure at the end of phase 2 prevents extracellular influx, while cytoplasmic Ca is pumped back into the SR by the SR Ca ATPase (SERCA). This drop in intracellular Ca concentration allows troponin and tropomyosin to reassume their inhibitory configurations, and myocardial relaxation ensues. SERCA activity is regulated principally by phospholamban, an SR membrane protein that, in its dephosphorylated state, inhibits Ca uptake by SERCA [10].
Extracellular calcium (Ca2+) enters through the sarcolemmal L-type calcium channels (LTCC), which in turn triggers Ca2+ release from the sarcoplasmic reticulum (SR) through the ryanodine receptors (RyR) known as calcium-induced calcium release (CICR) (Fig. 1). The increase in calcium concentration within the cardiomyocyte induces conformational changes in the actin myosin filaments resulting in contraction. Once contraction is over, the Ca2+ is pumped back into the SR by the SR Ca ATPase (SERCA) whose activity is regulated by phospholamban. Calcium is also returned extracellularly via the sodium-calcium exchanger (Fig. 1).
Sarcoplasmic Ca concentration determines the force of myocardial contraction. Higher intracellular levels lead to greater relief of troponin inhibition, rendering more actin–myosin complexes active. β-adrenergic stimulation augments cardiac contractility by increasing intracellular Ca influx via a G-protein (GS)/cAMP-mediated mechanism. The end-result is downstream protein kinase activation and phosphorylation of the LTCC, promoting Ca influx. β-agonism also promotes myocyte relaxation via phosphorylation of phospholamban, enhancing SERCA activity and preserving diastolic function [10]. Cholinergic stimulation, in turn, decreases contractility by downregulating intracellular Ca influx. An inhibitory G protein (GI) responding to acetylcholine transmission both inhibits adenylyl cyclase (decreasing cAMP levels) and opens myocyte potassium channels, hyperpolarizing the cell [10].
Nitric oxide
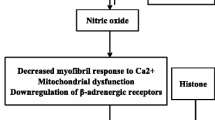
The effects of nitric oxide (NO) produced by nitric oxide synthase (NOS) have garnered heavy interest over the last 15 years as potential mediators of septic cardiomyopathy. NOS exist in three forms: NOS 1 and NOS 3 which are constitutive forms and as NOS 2 which is an inducible form [11]. Experiments in cultured cardiomyocytes of murine models show reduced β-adrenergic-induced inotropy upon exposure to activated immune cells [12]. Cytokine products of the activated cells include the enzyme NOS. Furthermore, NOS inhibitors such as NG-monomethyl-L-arginine (L-NMMA) block the negative inotropic effects of cytokines [13]. However, one illustrative experiment by Balligand et al. [14] showed the interplay of bacterial endotoxin, macrophage stimulation, NOS induction, cGMP production and reduced contractility. Here, rat macrophages were stimulated by exposure to lipopolysaccharide (LPS, a component of bacterial endotoxin) and then incubated with rat cardiomyocytes for 24 h. Amplitude of shortening of myocytes stimulated by β-agonists was significantly reduced by LPS exposure relative to controls, and this reduction was abolished by addition of the NOS inhibitor L-NMMA. Additionally, LPS exposure increased both nitrite production in myocytes and cGMP formation in reporter fibroblasts, suggesting a possible cGMP-mediated mechanism for NO-induced negative inotropy. Another experiment by Chung et al. [15] implicated inhibition of the adenylyl cyclase/cAMP second messenger system as the explanation for reduced contractility in the presence of NO. Peroxynitrite, a product of interaction between NO and the superoxide anion, has also been proposed as a possible cardiodepressant [2, 16].
Returning to our model of E-C coupling, GS stimulation by β-agonists ultimately leads to increased cAMP and phosphorylation of the LTCC, increasing Ca influx into myocytes. Cholinergic stimulation, alternatively, decreases cAMP levels while promoting cGMP-mediated myocardial relaxation. Though endothelial cells, myocytes, vascular smooth muscle cells, neurons and inflammatory cells all demonstrate NOS activity, it is not yet clear precisely how NO affects the cholinergic/adrenergic modulation of contractility.
Complicating the issue is the observation that cytokines exert cardioinhibitory effects in vitro over different time frames and both with and without NOS induction. Prolonged (>18 h) exposure to LPS, TNF-α, or IL-1β decreased contractility of guinea pig cardiomyocytes, while increasing cGMP content and NOS 2 levels [8]. All of these effects were abolished when the steroid dexamethasone was added to culture. Dexamethasone has been shown to inhibit induction of NOS 2 but not the activity of constitutively expressed NOS (NOS 1 or NOS 3). This seems to suggest that NO-mediated cardiodepression is dependent on the transcription and translation of an inducible protein. Yet we know from other cytokine studies that early myocyte depression and reduced intracellular Ca occur within minutes of exposure to IL-6 [17], and that TNF-α can block β-adrenergic effects on contractility without increasing mRNA for NOS 2 [18]. This implicates activation of constitutive NOS, NOS 1 and 3 as culprits in early septic myocardial depression, whereas inducible NOS 2 may mediate contractile depression that occurs in late sepsis [16].
The concept of NOS localization to subcellular domains helps clarify the complicated interplay between NO and the cardiovascular system. This paradigm proposes that: (1) NOS exists as various isoforms in each of the subcellular components in which it acts and (2) NO acts as a second messenger within these compartments via post-translational covalent nitrosylation of cysteine thiol groups [19]. By this theory, the NOS 3 isoform found in the sarcolemmal membrane produces NO that modifies the LTCC so as to inhibit Ca entry, promoting myocardial relaxation. Concomitantly, NO derived from NOS 1 in the SR nitrosylates RyR channel proteins in a manner that promotes Ca efflux from the SR, causing contraction [19]. NO signaling by nitrosylation is fast, reversible, and affected by the overall nitroso-redox balance of the system [20].
Nitric oxide (NO) produced by NOS 1 located in the sarcoplasmic reticulum (SR) acts on the ryanodine receptor (RyR) promotes calcium efflux (Ca2+) thereby promoting contraction. NO produced by NOS 3 in the sarcolemmal membrane acts on the L-type calcium channels (LTCC) to inhibit calcium entry thereby promoting myocardial relaxation (Fig. 3).

Localization of various isoforms of nitric oxide synthase
The oxidase enzymes NADPH oxidase and xanthine oxidase, as well as mitochondrial oxidative phosphorylation, generate the biologically relevant reactive oxygen species superoxide, hydrogen peroxide and the hydroxyl radical [21]. Reactive oxygen species (ROS) can be cytotoxic at high levels through a number of mechanisms. Aside from causing direct damage to DNA strands, proteins and lipids, ROS compete with NO for thiol substrates. Progressive oxidation of these substrates is irreversible, rendering them incapable of interacting with NO, thereby disrupting physiologic NO signaling [21]. Furthermore, NOS isoforms may directly interact with ROS-generating enzymes in a regulatory manner, such that NOS deficiency results in uncontrolled ROS production [19, 21]. Several animal models of heart failure have investigated the contributions of oxidative imbalance. Notably, Bendall et al. [22] showed mice deficient in a catalytic subunit of NADPH oxidase had less ventricular hypertrophy and interstitial fibrosis after exposure to angiotensin II relative to wild-type mice. Two other studies illustrated how inhibition of xanthine oxidase (XO) with allopurinol improves parameters of endothelial function in smokers and diabetics, two populations prone to atherosclerosis and resultant endothelial dysfunction [23, 24]. However, no trials in humans with CHF to date have shown that XO inhibition leads to significant improvements in symptoms, functional performance or clinical outcomes [21].
Intracellular energetics
Cardiac dysfunction in sepsis has been linked to impaired intracellular calcium homeostasis and disrupted high-energy phosphate production. In the cecal ligation and puncture (CLP) model of sepsis in the cat heart, cardiac sarcoplasmic reticulum shows a 46% reduction in the rate of ATP-dependent Ca uptake during hypodynamic sepsis [25]. The SR Ca-ATPase (SERCA) and its main regulatory protein, phospholamban (PL), control reuptake of Ca into the SR. cAMP-dependent protein kinases normally phosphorylate PL and activate SERCA. This implicates defective phosphorylation of SR proteins in the myocardial dysfunction of late sepsis [25].
Cytopathic hypoxia refers to the phenomenon of impaired mitochondrial oxygen consumption by septic muscle despite adequate oxygen supply, which leads to uncoupled oxidative phosphorylation and diminished ATP production [26]. Chen et al. [26] studied this phenomenon in late (>18 h) CLP-induced sepsis in rats and its susceptibility to “heat-shock” pre-treatment. They found decreased enzymatic activity of electron transport chain cytochrome c reductases, decreased expression of mitochondrial respiratory chain enzymes, and ultrastructural deformity in the mitochondria in the hearts of late sepsis rats. This translated to diminished ATP content in cardiomyocytes. Interestingly, all of the above septic-induced changes could be attenuated or reversed by whole-body heating rats to 41–42°C for 15 min prior to CLP. The group proposed heat-induced expression of “heat-shock proteins” (Hsp) as the protective mechanism, and demonstrated how the expression of 2 such proteins (Hsp 72 and Grp 75) was preserved in heat shock relative to control. Functionally, Hsp 72 helps transfer newly synthesized cytoplasmic proteins into mitochondria, while Grp 75 assists in mitochondrial protein folding and protein complex assembly [27, 28]. The group concluded that mitochondrial respiratory chain protein dysfunction correlates with clinical deterioration in sepsis, and may be alleviated by the heat-shock response.
Cellular adhesion molecules
Elaboration of TNF-α and IL-1 in LPS models of sepsis leads to neutrophil recruitment and inflammatory damage to both liver and lung tissue [29, 30]. Intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1) mediate neutrophil infiltration [31]. Raeburn et al. [31] demonstrated a 40% decrease in left ventricular developed pressure of isolated mice hearts 6 h after LPS injection, associated with temporal increases in expression of ICAM-1, VCAM-1 and myocardial neutrophils. Antibody blockade of either ICAM or VCAM abrogated contractile dysfunction, but neutrophil depletion by either antibody blockade or vinblastine pre-treatment did not. The authors suggested alteration in intracellular Ca fluxes and generation of oxygen-free radicals by antibody crosslinking of adhesion molecules as possible explanations that warrant further investigation.
Complement
The humoral immune response becomes activated in sepsis, triggering a cascade of complement proteins. Eventual cleavage of C5 generates C5a, a potent anaphylatoxin, and C5b, a component of the terminal membrane attack complex, which lyses bacterial membranes [1]. C5a is a powerful pro-inflammatory agent known to increase neutrophil chemotaxis, granular enzyme release, reactive oxygen species production, and synthesis of cardiodepressant cytokines [1]. Overproduction of C5a and its receptor (C5aR), which is expressed on cells in the heart, lungs, liver and kidneys, could contribute to the dysregulated immune response characteristic of sepsis and multi-organ dysfunction. Niederbichler et al. [32] examined the effects of C5a in CLP-induced sepsis on rat hearts in vivo and isolated cardiomyocytes in vitro. CLP resulted in significantly decreased left ventricular pressure in whole hearts, an effect reversed by administration of anti-C5a antibody (Ab) immediately after CLP. In isolated cardiomyocytes, exposure to recombinant C5a reduced contractility (measured as peak sarcomere shortening) in both CLP and sham-operated rats, but to a greater degree in CLP rats. There was a corresponding time-dependent increase in C5aR mRNA and protein extracted from CLP cardiomyocytes relative to sham cells. The authors proposed that constitutive expression of C5aR on cardiomyocytes mediates early C5a-induced cardiodepression, but sepsis enhances in vivo C5a production and C5aR expression on cardiomyocytes, resulting in delayed cardiomyopathy.
Other cytokine effects
Cytokines are peptides sporadically produced by nucleated cells in response to some stress or challenge [7]. Surgical insults, trauma, ischemia or sepsis can all induce the local release of these proteins from both somatic cells (endothelial, epithelial, fibroblasts) and immune cells (neutrophils, lymphocytes, macrophages). The cytokine signal serves as a language between the challenged cells [7]. This language is not determined by one or two peptides in isolation but rather by the confluent effects of circulating binding proteins, levels of cytokine receptor expression and the overall cytokine milieu.
As such, we have been largely unsuccessful in translating knowledge about cytokine behavior in vitro to efficacious therapeutic manipulations in vivo. For example, TNF-α and IL-1β are 2 potent proinflammatory cytokines released by macrophages in sepsis that have each demonstrated in vitro depression of cardiac contractility [33, 34]. A trial conducted in the early 1990s on 10 patients with refractory septic shock using a monoclonal TNF-α antibody (Ab) revealed a transient but statistically significant decrease in heart rate and increase in left ventricular stroke-work index [35]. Nevertheless, no large-scale clinical trials have shown a mortality benefit with TNF-α Ab in septic shock [4]. Similarly, a large phase III placebo-controlled trial of nearly 700 septic patients treated with IL-1 receptor antagonist failed to show any difference in 28-day mortality [36].
TNF-α may also trigger cardiac apoptosis in the LPS model of sepsis. Carlson et al. [37] examined TNF-α-dependent apoptotic pathways in isolated mouse cardiomyocytes that worked via activation of specific proteases termed capsases. They found LPS injection increased capsase-3 activity in cardiomyocytes, in association with pro-apoptotic patterns of gene expression and depressed left ventricular developed pressure in ex vivo hearts. TNF-α receptor knockout mice challenged with LPS exhibited markedly decreased capsase activity despite elevated TNF-α concentrations. Administration of the capsase-3 inhibitor zVAD in wild-type mice attenuated LPS-induced cardiodepression. However, LPS was still able to trigger some degree of decreased contractility in knockout mice, indicating the presence of TNF-α-independent mechanisms of contractile disturbance. It has been suggested that caspase-3 activation may be associated with decreased sensitivity of the myofilaments to calcium, or even structural breakdown of the sarcomere itself. Given the relative reversibility of septic cardiomyopathy, further studies are necessary to delineate the exact role of caspases and apoptosis.
Summary
Experimental models of sepsis show clear evidence of myocardial contractile disturbance both in vivo and in vitro. This disturbance is present even in early “hyperdynamic” shock, when aggressive volume replacement and adaptive left ventricular dilatation can combine to preserve cardiac output [38]. What exactly triggers septic cardiomyopathy is still unknown. As normal cardiac contractility depends on intracellular Ca fluxes and ATP-consuming reactions, we have proposed mechanisms based on impaired cellular Ca homeostasis and uncoupled oxidative phosphorylation. We have examined the interplay between cellular products upregulated in sepsis, such as cytokines and nitric oxide, and the adrenergic/cholinergic/G protein-mediated regulation of contractility. Several investigators have successfully manipulated components of the regulatory pathway to reverse septic cardiodepression on a cellular level in vitro, yet the results of large clinical trials targeting these components in humans with septic cardiomyopathy are disappointing. Initial studies with NOS inhibitors such as NMMA in patients with septic shock are conflicting. Even though peripheral circulatory failure was reversed by increasing vascular tone and which thereby reduced the requirement for pressors [39], use of NOS inhibitors has been associated with increased mortality [40]. Perhaps our models of sepsis fail to accurately portray the complex biological processes occurring in the live human [41]. Alternatively, we may not fully grasp the protective effects of the systemic inflammatory response we are attempting to modify [38]. Future advances in the management of septic shock will likely depend on our ability to reconcile adaptive physiologic responses with pathologic dysregulation of the immune response.
References
Hoesel LM, Niederbichler AD, Ward PA (2007) Complement-mediated molecular events in sepsis leading to heart failure. Mol Immunol 44:95–102
Hunter JD, Doddi M (2010) Sepsis and the heart. Br J Anaesth 104:3–11
Weisel RD, Vito L, Dennis RC, Hechtman HB (1977) Myocardial depression during sepsis. Am J Surg 133:512–521
Court O, Kumar A, Parillo JE, Kumar A (2002) Clinical review: myocardial depression in sepsis and septic shock. Critical Care 6:500–508
Parker MM, Suffredini AF, Natanson C, Ognibene FP, Shelhamer JH, Parrillo JE (1989) Responses of left ventricular function in survivors and nonsurvivors of septic shock. J Crit Care 4:19–25
Parker MM, Shelhamer JH, Bacharach SL et al (1984) Profound but reversible myocardial depression in patients with septic shock. Ann Int Med 100:483–490
Raeburn CD, Sheppard F, Barsness KA, Arya J, Harken AH (2002) Cytokines for surgeons. Am J Surg 183:268–273
Stein B, Frank P, Schmitz W, Scholz H, Thoenes M (1996) Endotoxin and cytokines induce direct cardiodepressive effects in mammalian cardiomyocytes via induction of nitric oxide synthase. J Mol Cell Cardiol 28:1631–1639
Pathan N, Hemingway CA, Alizadeh AA, Stephens AC, Boldrick JC, Oragui EE, McCabe C, Welch SB, Whitney A, O’Gara P, Nadel S, Relman DA, Harding SE, Levin M (2004) Role of interleukin 6 in myocardial dysfunction of meningococcal septic shock. Lancet 363:203–209
Malhotra R, Edelman ER, Lilly LS (2003) “Basic cardiac structure and function”. Pathophysiology of heart disease: a collaborative project of medical students and faculty. Lippincott Williams &Wilkins, Ed. Leonard Lilly. Philadelphia, PA, pp 10–26
Rudiger A, Singer M (2007) Mechanisms of sepsis-induced cardiac dysfunction. Crit Care Med 35(6):1599–1608
Gulick T, Pieper SJ, Murphy MA et al (1991) A new method for assessment of cultured cardiac myocyte contractility detects immune factor-mediated inhibition of β-adrenergic responses. Circulation 84:313–321
Hare JM, Colucci WS (1995) Role of nitric oxide in the regulation of myocardial function. Prog Cardiovasc Dis 38:155–166
Balligand J-L, Ungureanu D, Kelly RA et al (1993) Abnormal contractile function due to induction of nitric oxide synthesis in rat cardiac myocytes follows exposure to activated macrophage-conditioned medium. J Clin Invest 91:2314–2319
Chung MK, Gulick TS, Rotondo RE et al (1990) Mechanism of cytokine inhibition of β-adrenergic agonist stimulation of cyclic AMP in rat cardiac myocytes. Impairment of signal transduction. Circ Res 67:753–763
Zanotti-Cavazzoni SL, Hollenberg SM (2009) Cardiac dysfunction in severe sepsis and septic shock. Curr Opin Crit Care 15(5):392–397
Kinugawa K, Takahashi T, Kohmoto O, Yao A, Aoyagi T, Monomura S, Hirata Y, Serizawa T (1994) Nitric oxide-mediated effects of IL-6 on [Ca2+]i and cell contraction in cultured chick ventricular myocytes. Circ Res 75:285–295
Muller-Werdan U, Schumann H, Fuchs R, Reithmann C, Loppnow H, Koch S, Zimny-Arndt U, He C, Darmer D, Jungblut J, Stadler J, Holtz J, Werdan K (1997) Tumor necrosis factor α is cardiodepressant in pathophysiologically relevant concentrations without inducing nitric oxide synthase or triggering serious cytotoxicity. J Mol Cell Cardiol 29:2915–2923
Saraiva RM, Hare JM (2006) Nitric oxide signaling in the cardiovascular system: implications for heart failure. Curr Opin Cardiol 21:221–228
Xu L, Eu JP, Meissner G, Stamler JS (1998) Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science 279:234–237
Zimmet JM, Hare JM (2006) Nitroso-redox interactions in the cardiovascular system. Circulation 114:1531–1544
Bendall JK, Cave AC, Heymes C, Gall N, Shah AM (2002) Pivotal role of a gp91 (phox)-containing NADPH oxidase in angiotensin II-induced cardiac hypertrophy in mice. Circulation 105:293–296
Butler R, Morris AD, Belch JJ, Hill A, Struthers AD (2000) Allopurinol normalizes endothelial dysfunction in type 2 diabetics with mild hypertension. Hypertension 35:746–751
Guthikonda S, Sinkey C, Barenz T, Haynes WG (2003) Xanthine oxidase inhibition reverses endothelial dysfunction in heavy smokers. Circulation 107:416–421
Wu LL, Ji Y, Dong LW, Liu MS (2001) Calcium uptake by the sarcoplasmic reticulum is impaired during the hypodynamic phase of sepsis in the rat heart. Shock 15:49–55
Chen HW, Hsu C, Lu TS, Wang SJ, Yang RC (2003) Heat shock pretreatment prevents cardiac mitochondrial dysfunction during sepsis. Shock 20:274–279
Stuart RA, Cyr DM, Neupert W (1994) Hsp70 in mitochondrial biogenesis: from chaperoning nascent polypeptide chains to facilitation of protein degradation. Experientia 50:1002–1011
Mizzen LA, Kabiling AN, Welch WJ (1991) The two mammalian mitochondrial stress proteins, grp 75 and hsp 58, transiently interact with newly-synthesized mitochondrial proteins. Cell Regul 2:165–179
Hewett JA, Schultze AE, VanCise S, Roth RA (1992) Neutrophil depletion protects against liver injury from bacterial endotoxin. Lab Invest 66:347–361
Tsuji C, Minhaz MU, Shioya S, Fukahori M, Tanigaki T, Nakazawa H (1998) The importance of polymorphonuclear leukocytes in lipopolysaccharide-induced superoxide anion production and lung injury: ex vivo observation in rat lungs. Lung 176:1–13
Raeburn CD, Calkins CM, Zimmerman MA, Song Y, Ao L, Banerjee A, Harken AH, Meng X (2002) ICAM-1 and VCAM-1 mediate endotoxemic myocardial dysfunction independent of neutrophil accumulation. Am J Physiol Regul Integr Comp Physiol 283:R477–R486
Niederbichler AD, Hoesel LM, Westfall MV, Gao H, Ipaktchi KR, Sun L, Zetoune FS, Su GL, Arbabi S, Sarma JV, Wang SC, Hemmila MR, Ward PA (2006) An essential role for complement C5a in the pathogenesis of septic cardiac dysfunction. J Exp Med 203:53–61
Gu M, Bose R, Bose D, Yang J, Li X, Light RB, Mink S (1998) Tumor necrosis factor-alpha but not septic plasma depresses cardiac myofilament contraction. Can J Anesth 45:280–286
Hosenpud JD, Campbell SM, Mendelson DJ (1989) Interleukin-1 induced myocardial depression in an isolated beating heart preparation. J Heart Transplant 8:460–464
Vincent JL, Bakker J, Marecaux G, Schandene L, Kahn RJ, Dupont E (1992) Administration of anti-TNF antibody improves left ventricular function in septic shock patients: results of a pilot study. Chest 101:810–815
Opal SM, Fisher CJ Jr, Dhainaut JF, Vincent JL, Brase R, Lowry SF, Sadoff JC, Slotman GJ, Levy H, Balk RA, Shelly MP, Pribble JP, Labrecque JF, Lookabaugh J, Donovan H, Dubin H, Baughman R, Norman J, DeMaria E, Matzel K, Abraham E, Seneff M (1997) Confirmatory interleukin-1 receptor antagonist trial in severe sepsis: a phase III, randomized, double blind, placebo-controlled multicenter trial. The interleukin-1 receptor antagonist sepsis investigator group. Crit Care Med 25:1115–1124
Carlson DL, Willis MS, White J, Horton JW, Giroir BP (2005) Tumor necrosis factor-α-induced caspase activation mediates endotoxin-related cardiac dysfunction. Crit Care Med 33:1021–1028
Grocott-Mason RM, Shah AM (1998) Cardiac dysfunction in sepsis: new theories and clinical implications. Intensive Care Med 24:286–295
Watson D, Grover R, Anzueto A, Lorente J, Smithies M, Bellomo R, Guntupalli K, Grossman S, Donaldson J, Le Gall JR (2004) Cardiovascular effects of the nitric oxide synthase inhibitor NG-methyl-L-arginine hydrochloride (546C88) in patients with septic shock: Results of a randomized, double-blind, placebo-controlled multicenter study (study no. 144-002). Crit Care Med 32(1):13–20
Lopez A, Lorente JA, Steingrub J, Bakker J, McLuckie A, Willatts S, Brockway M, Anzueto A, Holzapfel L, Breen D, Silverman MS, Takala J, Donaldson J, Arneson C, Grove G, Grossman S, Grover R (2004) Multiple-center, randomized, placebo-controlled, double-blind study of the nitric oxide synthase inhibitor 546C88: effect on survival in patients with septic shock*. Crit Care Med 32(1):21–30
Levy RJ, Deutschman CS (2004) Evaluating myocardial depression in sepsis. Shock 22:1–10
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Flynn, A., Chokkalingam Mani, B. & Mather, P.J. Sepsis-induced cardiomyopathy: a review of pathophysiologic mechanisms. Heart Fail Rev 15, 605–611 (2010). https://doi.org/10.1007/s10741-010-9176-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-010-9176-4