
Abstract
Infarct size is determined not only by the duration and severity of ischemia, but also by pathological processes initiated at reperfusion (reperfusion injury). Numerous pharmacological strategies have been reported which administer drugs at or just before the onset of reperfusion, with subsequent salubrious effects, notably a reduction in infarct size. However, few if any of these strategies have become standard of care in the catheterization laboratory setting. Postconditioning, defined as repeated brief cycles of reperfusion interrupted by ischemia (or hypoxia) applied at the onset of reperfusion, was recently introduced as a mechanical strategy to attenuate reperfusion injury. Postconditioning intervenes only during the first few minutes of reperfusion. However, it reduces endothelial activation and dysfunction, the inflammatory response to reperfusion, necrosis, and apoptosis both acutely and long-term. Cardioprotection has been demonstrated by multiple independent laboratories and in multiple species. Postconditioning stimulates G-protein coupled receptors by their cognate endogenously released ligands and surprisingly activates survival kinases that may converge on mitochondrial KATP channels and the permeability transition pore. Postconditioning has been shown in two clinical studies to reduce infarct size in patients undergoing percutaneous coronary intervention in the catheterization laboratory, and at least five other studies are in some phase of implementation. This significant reduction in infarct size has implications for reduction in heart failure as a consequence of myocardial infarction, but this link has yet to be demonstrated. The salubrious effects of postconditioning are an indirect validation of the experimental and clinical importance of reperfusion injury in the setting of coronary artery occlusion.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cardiovascular disease is the leading cause of death in both men and women in the United States, accounting for approximately 40% of all deaths in 2000. This percentage is greater than the next leading causes of death combined (cancer, chronic respiratory disease, accidents, diabetes mellitus, and influenza/pneumonia). About 5% of patients with coronary heart disease die of heart failure. The incidence of heart failure as estimated by hospital discharges has increased approximately 145% between 1970 and 2000. Currently, it is estimated that between 2.5 and 3 million people in the United States (representing 1% of the population) has heart failure, with over half a million new cases being diagnosed each year [1–3]. In 1993, the 5 year survival after the diagnosis of heart failure was as low as 25–35% [4], but these mortality statistics have improved with the use of ACE inhibitors and beta blockers. Treatment for heart failure accounts for over 6% of the total health care expenditures.
There are a number of pathological conditions that contribute to the etiology of heart failure. Hypertension is a strong independent risk factor. Myocardial infarction is likewise an independent risk factor for heart failure. In 2005, approximately 1.2 million Americans suffered a myocardial infarction, of which 42% died. After myocardial infarction, the surviving myocardium undergoes a complex sequence of remodeling changes that present functioning and hemodynamic advantages in the short-term, but which ultimately cause heart failure in the long-term. In experimental studies, the degree of this deleterious remodeling is proportional to the size of the infarct [5] which causally links the development of heart failure to myocardial infarction. Indeed, permanent or transient coronary artery occlusion is a popular experimental model of heart failure [6]. Accordingly, clinical reports indicate that infarct size estimated by peak plasma creatine kinase activity is an independent predictor of LV remodeling and hence heart failure [7].
In view of the important role that infarct size plays in the etiology of heart failure, limiting infarct size is an important strategy to reduce the incidence and severity of heart failure, in addition to other therapeutic strategies that target the remodeling process (i.e., β-adrenergic inhibitors, angiotensin-converting enzyme inhibitors, angiotensin receptor blockers). It appears likely that the limitation of unfavorable LV remodeling is a physiological consequence of infarct size limitation [8], potentially through a decrease in the untoward remodeling and geometric changes (dilatation) leading to heart failure. Therefore, cardioprotective strategies that reduce infarct size achieve not only short-term benefits, but may have long-term benefits by reducing the human and financial burden of heart failure.
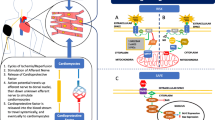
Postconditioning is defined as repeated brief cycles of reperfusion interrupted by ischemia (or acidosis) applied at the onset of reperfusion [9]. In its brief lifetime since 2003 [9], postconditioning has enjoyed a steep trajectory that has taken it from an unlikely bench top experimental “curiosity” to a clinically validated strategy that significantly reduced infarct size in patients undergoing percutaneous coronary interventions (PCI) [10–12]. In addition, at the time of this writing, there are five clinical trials either enrolling patients or ready for launch, all outside the United States, and one on “pharmacological” postconditioning with adenosine for ST-segment elevation myocardial infarction (STEMI).
Postconditioning is undoubtedly a form of modified reperfusion, and in a broad sense it may be similar to other mechanical interventions that mechanically alter the onset of reperfusion [13], such as gradual or controlled reperfusion [14, 15]. However, it has not been shown that gradual reperfusion and postconditioning exert a similar degree of infarct reduction, or similar vasculoprotection to coronary vascular endothelium, or whether the molecular mechanisms are involved. By its time frame of exerting cardioprotection within the first few minutes of reperfusion [16] postconditioning interrupts events that are occurring within the first few minutes of reperfusion—events that obviously play a causal role in postischemic injury. As Piper and associates [17, 18] and others [14, 19] have stated, the first few minutes of reperfusion are very active indeed in contributing to the pathophysiology of not only myocardial necrosis, but also to endothelial cell dysfunction and apoptosis. A summary of these early events is shown in Table 1. The attenuation of postischemic injury by postconditioning is an indirect and very compelling validation of the existence of reperfusion injury, not only in experimental models, but also in patients undergoing evolving myocardial infarction and clinically destined for angioplasty [10–12].
Protection against acute myocardial infarction
The initial study by Zhao et al. [9] in the canine model of coronary artery occlusion established that postconditioning significantly reduced infarct size. The ability to reduce infarct size has been confirmed by multiple labs and in multiple species, including mouse [20, 21], rat [22–24], rabbit [25–27], canine [9, 28], and pig [29] (but not without some controversy [30]). However, its protection appears to go beyond protecting only myocytes from necrosis; postconditioning also reduces apoptosis [31, 32]. Postconditioning may reduce pro-apoptotic triggers such as the generation of oxygen radicals and accumulation of calcium in mitochondria. However, the vascular endothelium is also spared by postconditioning. It is the opinion of the authors and other investigators that the coronary vascular endothelium is a critical factor in the initiation of the inflammatory response to reperfusion, and as such preservation of the coronary vascular endothelium is causally related to a reduction in infarct size, as has been reviewed elsewhere [33–36]. The remainder of the review will discuss the mechanisms by which postconditioning reduces necrosis and apoptosis. The observation that necrosis and infarct size are determinants of heart failure, and apoptosis is an important mechanism leading to heart failure makes postconditioning a potential therapeutic strategy that may have a future impact on the incidence and severity of heart failure secondary to acute myocardial infarction. Indeed, a recent study by Zhu et al. [24] reported that postconditioning reduced infarct size (TTC staining and lactate dehydrogenase activity) and improved function in hearts with chronic (6 weeks) myocardial hypertrophy induced by permanent coronary artery ligation or by one kidney/one clip hypertension. After the chronic condition each heart was subjected to 40 min global ischemia (Langendorff apparatus) and 90 min of reperfusion with or without postconditioning following an algorithm of 10 sec reperfusion and 10 sec complete ischemia repeated for 6 cycles. The infarct-sparing effect of postconditioning was comparable between the non-remodeled (“healthy”) hearts and the two hypertrophy models. However, functional recovery after global ischemia was somewhat less (but still significantly greater than controls) in the one clip/one kidney model than in the infarcted or non-remodeled cohorts.
Mechanical attenuation of reperfusion
A proximal-most mechanism of postconditioning may be the slow reintroduction of oxygen and persistence of tissue acidosis. In elegant experiments performed in regionally ischemic isolated perfused rabbit hearts by Cohen et al. [37], the cardioprotection exerted by postconditioning was replicated by reperfusion with hypercapnic buffer (pH was 6.9) for the first 2 min of reflow. This protection with acidotic perfusate was abrogated by an alkalotic (pH 7.7) buffer, and by the oxygen radical scavenger N-2-mercaptopropionyl glycine. These data suggest that maintaining tissue acidosis for the first 2 min of reperfusion is an important mechanism in postconditioning. Maintaining tissue acidosis may attenuate opening of the mitochondrial permeability transition pore (mPTP), while the gradual reintroduction of oxygen may generate lower levels of oxygen radicals for tissue signaling rather than the large levels associated with tissue injury. More experimentation is needed to confirm this involvement of tissue acidosis by measuring tissue pH and acid products during the short window of postconditioning, and the relationship between tissue pH and the mPTP, and signaling versus cardiodestructive levels of reactive oxygen species. In addition, more research has to determine the relationship between maintaining tissue acidosis, the gradual reintroduction of oxygen and endogenous ligands that trigger postconditioning (adenosine, bradykinin, opioids). Whether these mechanisms operate independently and some operate in an integrated manner must still be demonstrated.
Inhibition of endothelial dysfunction, neutrophil actions and the inflammatory response to reperfusion
A number of investigators have suggested that some of the early events initiated at reperfusion are similar to an inflammatory-like response [38–42]. There is ample evidence in the experimental literature to support an inflammatory-like response that begins in the very early moments of reperfusion with activation of neutrophils and vascular endothelial cells in the reperfused area, and a subsequent recruitment of neutrophils to first adhere to the endothelium (at first loosely with rolling along the surface, later firm adherence) and then finally migrate into the parenchyma (>4 h after onset of reperfusion). Both neutrophils and the vascular endothelium elaborate pro-inflammatory mediators (TNFα, IL-6, IL-8) and oxidant species that can damage the endothelium in these early minutes [19, 36], and finally damage cardiomyocytes. These same oxidants also neutralize constitutively expressed nitric oxide and form the potent oxidant peroxynitrite. The hallmarks of endothelial dysfunction are: impaired vasorelaxation to acetylcholine, and increased adherence of neutrophils, both related to impaired release of nitric oxide secondary to dysfunction of the endothelial receptors used to stimulate relaxation, a loss of cofactors such as tetrahydrobiopterin, or damage to the eNOS apparatus itself.
Postconditioning has been shown to attenuate endothelial dysfunction, reduce neutrophil adherence to endothelium and their accumulation in the reperfused myocardium, and pro-inflammatory mediators such as TNFα. The importance of neutrophils in reperfusion injury has been disputed by some [43]. Indeed, the persistence of cardioprotection is observed in cell-free model systems which would appear to argue against an active anti-inflammatory component to postconditioning cardioprotection. Preliminary unpublished data from our laboratory (A. Granfeldt Petersen et al.) using neutrophil antiserum to deplete the neutrophil population in an in vivo rodent model of coronary artery occlusion-reperfusion show that both neutrophil depletion and postconditioning separately reduce infarct size to the same extent (∼28%), but there was no significant further reduction in infarct size when postconditioning and neutrophil depletion were combined. Whether neutrophils as well as oxidants and pro-inflammatory mediators contribute significantly to the pathogenesis of reperfusion injury, as well as their role in postconditioning, is on on-going and vigorous debate.
G-protein coupled receptors: a possible convergence of triggers of postconditioning
G-protein coupled receptors (GPCR) have recently been implicated in cardioprotection at a very proximal point—the cell membrane. The stimulation of the GPCR for adenosine [44–46], bradykinin [47, 48], and opioids at the time of reperfusion have been associated with a reduction of infarct size. In addition, there may be considerable cross-talk between these receptors, e.g., stimulation of the adenosine receptor by opioid agonists and vice verse [49]. Involvement of three GPCRs in postconditioning is reviewed below. Whether blockade at the G-protein transduction level blocks the effects of these ligands has not been attempted. However, it is interesting that blockade of either adenosine or opioid receptors completely blocks postconditioning, suggesting an all-or-none response, rather than demonstrating a graded or incremental reversal of infarct reduction. In addition, a combination of these GPCR agonists has not been administered to determine if there is an additive or synergistic effect.
Adenosine in postconditioning
Of the four adenosine receptor subtypes (A1, A2A, A2B, A3), the A2A and A3 receptors have been shown to be cardioprotective when activated at reperfusion [46, 50]. Recently, Yang et al. [51] reported that the selective A2A adenosine receptor agonist ATL 146e administered intraperitoneally 2 min before reperfusion reduced infarct size in wild-type mice, but had no effect in global A2A receptor knock-out mice, or chimeric mice in which the A2A receptor was not expressed in bone marrow derived cells. These data support the concept that adenosine acts to attenuate reperfusion injury by an anti-inflammatory mechanism. This study [51] also showed in RAG-1 knock-out mice that inhibition of T lymphocytes may be a mechanism of action, consistent with the notion that T lymphocytes (perhaps CD4+) may generate the cytokines and chemokines that recruit neutrophils to the reperfused myocardium. This anti-inflammatory effect of A2A adenosine receptor stimulation during postconditioning is consistent with the following observations: (1) adenosine levels are elevated during ischemia, and the washout of adenosine is delayed by postconditioning [20]; (2) non-selective inhibition of adenosine receptors [20, 25] and the A2A receptor specifically [20], abrogates the cardioprotection of postconditioning; (3) the reduction of neutrophil adherence, accumulation, and preservation of the vascular endothelium are consistent with the physiological effects of adenosine in models of ischemia-reperfusion. Recently, Philipp et al. [26] from Cohen’s and Downey’s group have suggested that postconditioning activates the A2B receptor and PKC rather than the A2A receptor. The purported A2B antagonist MRS 1754 blocked the infarct-sparing effect of postconditioning. Studies have been limited to pharmacological blockade, and lack confirmation in appropriate A2B knock-out or knock-down (siRNA against A2B mRNA) models. Further studies are needed in this area to clarify which receptor subtype(s) are involved in the endogenous adenosine-mediated protection of postconditioning.
Bradykinin
Yang et al. [48] reported that bradykinin administered at reperfusion reduced infarct size by mechanisms that signaled through the reperfusion injury survival kinase pathway and nitric oxide production. Similar results were reported by Bell and Yellon in a murine model of ischemia-reperfusion [47]. There is no formal publication on whether endogenous bradykinin is involved in postconditioning.
Opioids in postconditioning
There are three opioid receptor subtypes, the μ, δ, and κ. The δ and κ receptors are expressed in cardiomyocytes and coupled to Gi. The myocardium is able to synthesize the three major endogenous opioid peptides (enkephalins, endorphins, and dynorphins). The release of opioid peptides and subsequent stimulation of κ and δ receptors has been implicated in the cardioprotection of preconditioning [52]. Recent evidence has suggested that exogenous opioids administered at reperfusion are cardioprotective [53, 54]. Accordingly, the non-selective opioid morphine, or the δ opioid receptor agonists BW373U86 and fentanyl isothiocyanate administered at reperfusion, reduce infarct size comparable to that observed with a pretreatment administration in the rat model of coronary artery occlusion and reperfusion [53]. However, κ opioid receptor stimulation at reperfusion was not cardioprotective. The mechanism of this opioid-induced infarct salvage was suggested to be activation of the RISK pathway components PI-3 kinase, mammalian target of rapamycin (mTOR), p70s6 kinase, and phosphorylation and subsequent inactivation of GSK-3β. Hence activation of opioid receptors at reperfusion is cardioprotective; a cardioprotective role for endogenous opioids is also suggested by the antagonist data.
Since opioid precursors and peptides may be synthesized and released in myocardium during ischemia/reperfusion, the hypothesis may be put forward that opioids mediate in part the infarct sparing effect of postconditioning. Kin et al. [55] demonstrated the involvement of endogenous opioid peptides in postconditioning. In the rat model of 30 min coronary artery occlusion followed by 3 h of reperfusion, postconditioning cardioprotection was completely abrogated by the non-subtype selective antagonist naloxone and its peripherally restricted quaternary derivative naloxone methiodide administered 5 min before reperfusion. Blockade of either the δ receptor (naltrindole hydrochloride) or the κ receptor (nor-binaltorphimine dihydrochloride) abrogated postconditioning as well. Whether μ receptor stimulation was involved was not clear. Hence, in contrast to preconditioning, in which only the δ receptor is suspected of being involved, both the κ and δ opioid receptors may be involved in postconditioning. Further studies by Zatta et al. (unpublished) show that postconditioning increases precursor proenkephalin levels in area at risk myocardium, and achieves a normalization of active enkephalin levels without effecting methionine enkaphalin or methionine-enkephalin-arginine-phenylalanin, in contrast to preconditioning which preserves active enkephalin metabolites. The increased enkephalin pool may allow an increased stimulation of opioid receptors, or a sustained release over an undetermined period of reperfusion.
Signaling in postconditioning cardioprotection
PI3-K and ERK ½
Surprisingly, the very short period of intervention that defines postconditioning activates a complex and growing array of molecular pathways. This network of pathways has been reviewed comprehensively by Hausenloy and Yellon [56]. The reperfusion injury survival kinases (RISK) [56, 57] PI3 kinase and MEK-ERK1/2 are distal targets of GPCR activation, so activation of these kinase pathways would be consistent with the involvement of GPCR ligands. Both pathways are stimulated at early reperfusion, placing this timing within the temporal frame of postconditioning. The PI3 kinase/Akt pathway likely is proximal to endothelial nitric oxide synthase [56], the activation of which has been found to be important to cardioprotection by postconditioning [27], and involves two targets that have been shown to be pivotal to postconditioning cardioprotection—the mitochondrial KATP channel [27, 58] and the mitochondrial permeability transition pore m(PTP) [59]. The mPTP is a potential “switch” not only of cell survival or death, but of pursuit along the necrotic or apoptotic pathways. Inhibition of the PI3 kinase pathway has, in most studies, abrogated infarct reduction by postconditioning [23, 25]. Likewise, the MEK-ERK1/2 pathway has also been reported to be an important pathway engaged by postconditioning [27, 60]. Interestingly, both preconditioning and postconditioning may have these two pathways in common. The involvement of these two pathways is entirely consistent with the importance of GPCR stimulation by endogenous ligands such as adenosine and opioids.
Protein kinase C (PKC)
PKC is a key signaling pathway in cardioprotection by preconditioning [61]. GPCR ligands such as adenosine and opioid agonists stimulate phospholipase C and the generation of inositol triphosphate and diacyglycerol, the latter of which stimulates PKC. In rodents, PKCɛ and δ are the predominant isoforms involved in preconditioning [62] and postconditioning, whereas PKCα is important in cardioprotection by preconditioning in pigs [63]. PKCδ may be involved in the pathogenesis of myocardial injury after ischemia-reperfusion [64, 65]. This isoform translocates to mitochondria within the first 5 min of reperfusion, which is within the time frame of the postconditioning effect. PKCδ is associated with pathophysiological events including increased superoxide anion generation, mitochondrial dysfunction and the release of cytochrome c and downstream pro-apoptotic factors [66, 67]. On the other hand, PKCɛ is associated with cardioprotection [68, 69] potentially by inhibiting the mitochondrial permeability transition pore [70]. Zatta et al. [71] recently reported that the infarct sparing effect of postconditioning in a rat model was abrogated by the non-selective PKC antagonist chelerythrine administered 5 min before the onset of reperfusion. Similar results were also reported by Penna et al. [72]. In addition, Zatta et al. [71] also showed that infarct size reduction by postconditioning was reversed by the PKCɛ specific inhibitor KIE1-1. Inhibition of PKCδ with rottlerin given 5 min before reperfusion by itself reduced infarct size, but did not alter (i.e., further reduce) the infarct size reduction achieved with postconditioning (Fig. 1). Postconditioning increased total cell homogenate levels of phosphorylated PKCɛ relative to the decreased levels observed after ischemia-reperfusion, suggesting a translocation site other than the mitochondrion, while phosphorylated levels and mitochondrial translocation of PKCδ were reduced. These data suggest that postconditioning cardioprotection is dependent on PKC signaling, and the mechanical maneuver increases the cardioprotective component of endogenous PKC signaling (PKCɛ) while simultaneously decreasing the cardio-destructive component (PKCδ). Philipp et al. [26] have also reported that cardioprotection by postconditioning is dependent on PKC signaling since the non-selective PKC inhibitor chelerythrine given 5 min before reperfusion aborted the infarct sparing effect of postconditioning in an in vivo rabbit model of coronary artery occlusion and postconditioning (4 cycles of 30 sec reperfusion/ischemia). A signaling role for PKC is consistent with the involvement of GPCR activators of PKC, specifically endogenous adenosine [20] and opioids.

The effects of PKC inhibitors on infarct size (area of necrosis/area at risk mass) reduction by postconditioning (postcon) in the rat model of 30 min coronary artery occlusion and 3 h of reperfusion. (A) Effects of the non-selective PKC inhibitor chelerythrine (5 mg/kg) administered intravenously 5 min before reperfusion or the PKCɛ inhibitor KIE1-1 (3.8 mg/kg). (B) The effects of the PKCδ inhibitor rottlerin (0.3 mg/kg body weight) alone or given before postconditioning. Values are mean ± standard errors of the mean. *P < 0.05 versus control; †P < 0.05 versus postconditioning group (From Zatta et al. [71] with permission)
Postconditioning and reactive oxygen species (ROS)
Reactive oxygen species include the superoxide anion and its products (H2O2 and •OH), and nitric oxide and its products (especially ONOO-). Under normal conditions oxidants are generated during the reduction of oxygen to water in mitochondria. Oxidants are part of cellular homeostasis, signaling, differentiation, mitosis, and immune responses. On the other hand, ROS are also an integral response to stresses such as ischemia-reperfusion. Initially described as the “oxygen paradox” by Hearse et al. [73] a robust generation of ROS has been shown to occur at reperfusion in myocardium [74, 75] and in isolated mitochondria [76]. In addition, the direct administration of ROS in concentrations approximating those observed at reperfusion causes injury to myocardium [77]. ROS are elaborated by cardiomyocytes, coronary vascular endothelium and inflammatory cells such as neutrophils. ROS cause injury by oxidation of lipid peroxidation of membranes, protein denaturation, and by causing breaks in genomic DNA. Hence, ROS play a dual role: that of signaling molecule and injury mechanism. This dual role of ROS may be based on several factors related to (1) the concentration achieved locally; (2) conversion of less reactive species into more potent species; (3) the environment favoring release of iron to fuel Fenton chemistry; and (4) the activation or shift of key ROS-generating enzymes. For example, the normally active xanthine dehydrogenase shifts to xanthine oxidase under ischemic conditions. In addition, cytokines and thrombin released at reperfusion can activate endothelial NADPH oxidase.
Preconditioning is mediated, in part, by elaboration of superoxide anions, possibly from mitochondria via signaling cascades that involve GPCR, PKC and KATP channels. Conversely, postconditioning may lead directly or indirectly to a reduction in ROS, which may be a factor differentiating the two cardioprotective strategies. Sun et al. [78] demonstrated that hypoxic postconditioning—the in vitro simulation of “ischemic” postconditioning—in cultured neonatal rat cardiomyocytes reduced ROS generation (superoxide anions and H2O2), which was associated also with attenuated cytosolic and mitochondrial calcium loading, and reduced cell death. In in vivo models of coronary artery occlusion-reperfusion, postconditioning was associated with less dihydroethidium-positive signal from oxidant-generating cells in the ischemic-reperfused myocardium [16, 28]. These observations are consistent with the reduction in lipid peroxidation products reported by several investigators in in vivo models of coronary artery occlusion and reperfusion [9, 16, 28]. In addition, postconditioning attenuated superoxide generation from the coronary vascular endothelium in the reperfused area after either 3 h or 24 h of reperfusion [79]. This reduction in endothelial ROS generation may be, in part, responsible for the preservation of endothelial function of epicardial coronary arteries observed in in vivo postconditioned hearts [9]. These data would suggest that postconditioning reduces oxidant-induced injury. However, Penna et al. [72] demonstrated in isolated rat hearts that the scavenger N-acetyl cysteine (NAC) given at reperfusion abrogated the infarct reduction by postconditioning. In addition, delaying administration of NAC until after the postconditioning algorithm was applied did not abolish the maneuver’s protection. These data suggest that the mechanisms of postconditioning cardioprotection may involve both preservation of the ROS-related signaling pathways and a reduction in oxidant-induced injury. Further research is necessary to clarify this very interesting duality. Postconditioning may also preserve antioxidant reserves in previously ischemic myocardium. Serviddio et al. [76] reported that postconditioning attenuated the oxidation of glutathione (GSH), the major intracellular antioxidant system. Hence, a reduction in the ROS burden may not only prevent direct tissue injury, but may preserve endogenous antioxidant levels in the myocardium.
The mitochondrial transition pore
The mitochondrial permeability transition pore (mPTP) is a voltage-dependent pore localized in the inner mitochondrial membrane. In the normally closed position, the mPTP impedes the movement of small molecules <1500 Da and water into the mitochondrial matrix. Opening of this pore allows transfer of small molecules into the matrix exacerbated by the osmotic properties of the matrix proteins. The inner membrane is capable of sustaining increases in matrix volume because of the convolutions of the cristae. However, the outer membrane is more vulnerable to such swelling, and will rupture, with a resulting collapse of the transmembrane potential, loss of ATP synthetic capacity, and release of pro-apoptotic factors such as cytochrome c. Key pathophysiological features of reperfusion conspire to open the mPTP at the onset of reperfusion, with the resultant accumulation of calcium and oxidants, and alkalinization of intracellular pH (acidosis impedes mPTP opening). Accordingly, opening of the mPTP has been shown to occur at reperfusion [80], notably within the first few minutes [81], which again places this event within the postconditioning window. Inhibition of mPTP opening with cyclosporine A, sanghliferin A or NIM811 reduces cell death and infarct size. After in vivo postconditioning, Argaud et al. [59] demonstrated that mitochondria isolated from the ischemic-reperfused myocardium were resilient to Ca2+-induced mPTP opening similar to that achieved by the mPTP opening inhibitor NIM811. These mitochondrial responses were associated with better preserved morphology. There are still questions whether the harvested mitochondria represented a more robust population in the postconditioning group versus the control group, and the comparison to NIM811 is indirect evidence only that attenuation of mPTP is causally linked to cardioprotection. However, these data are suggestive and present a good spring board for more definitive studies on the causal relationship of mPTP status and cardioprotection with postconditioning. A subsequent study demonstrated that attenuation of mPTP was associated with phosphorylation of Akt [82]. However, whether protection from mPTP opening by postconditioning is localized to cardiomyocytes or other cells involved in reperfusion injury is not known.
Postconditioning in hearts with co-morbidities
A preliminary report by Donato et al. [83] showed that postconditioning reduced infarct size and improved functional recovery following 30 min of global ischemia (isolated perfused preparation) and 30 min of reperfusion with or without postconditioning (2 cycles of 30 sec reperfusion/30 sec ischemia). A full-length publication has not appeared from these authors. In contrast, Iliodromitis et al. [84] reported that postconditioning did not reduce infarct size in a rabbit model of chronic (6 weeks) hypercholesterolemia with atherosclerosis, whereas the cardioprotection of preconditioning persisted. As discussed above, postconditioning cardioprotection was shown by Zhu et al. [24] to persist in two rodent models of myocardial remodeling (myocardial infarction, hypertension). Whether postconditioning efficacy is lost in diabetes or older hearts is not known as yet. Therefore, there are some experimental data that suggest postconditioning may be of limited value in models with isolated co-morbidities. However, these experimental data are inconsistent with the salubrious effects of postconditioning in patients undergoing percutaneous coronary intervention (PCI) in the catheterization laboratory. In the study by Staat et al. [10] the patients undergoing postconditioning had demographics similar to those treated by standard PCI, and consistent with the normal population of patients undergoing such procedures: mean age was 58 ± 4 years, 38% had high blood pressure, 57% were smokers, 80% had dyslipidemia, and 20% had diabetes. These patients were therefore “models” of multiple risk factors. In the study by Darling et al. [11] which also reported reduction in infarct size in patients treated by postconditioning (multiple balloon inflations), there were similar co-morbidities in the postconditioned group as in the standard PCI group. Therefore, although experimental models may identify limitations of one cardioprotective strategy or another, the translation of these results to the clinical scenario has to be done with appropriate caution and validation.
Concluding remarks
The concept that reperfusion is essential for myocardial salvage is abundantly justified experimentally and clinically. However, the concepts that (1) reperfusion is a component of the “wavefront” pattern of injury and (2) reperfusion as an opportunity to apply cardioprotective therapeutics to reduce postischemic injury and infarct size has enjoyed limited appreciation for some decades [85, 86], and has not been given due attention until very recently [13, 17, 18, 40]. Although hundreds of studies have investigated cardioprotective strategies applied at reperfusion, few if any have been adopted into clinical practice [87]. Postconditioning has highlighted the importance of the first few minutes of reperfusion in determining infarct size even 24 h or more after reperfusion [12, 59, 79]. In addition, postconditioning has been used in limited but growing numbers of clinical trials after only 3 short years since its introduction. It is hoped that this and other strategies aimed at attenuating post-ischemic injury by applying a cardioprotective strategy at reperfusion will be tested in clinical studies, and if appropriate, be adopted into the armamentarium of reperfusion therapeutics for patients undergoing PCI.
References
Gheorghiade M, Bonow RO (1998) Chronic heart failure in the United States. A manifestation of coronary artery disease. Circulation 97:282–289
Massie BM, Shah NB (1997) Evolving trends in the epidemiologic factors of heart failure: rational for preventive strategies and comprehensive disease management. Am Heart J 133:703–712
Sharpe N, Doughty R (1998) Epidemiology of heart failure and ventricular dysfunction. Lancet 352:3–7
Ho KK, Anderson KM, Kannel WB, Grossman W, Levy D (1993) Survival after the onset of congestive heart failure in Framingham heart study subjects. Circulation 88:107–115
Pfeffer MA, Braunwald E (1990) Ventricular remodeling after myocardial infarction. Experimental observation and clinical implications. Circulation 81:1161–1172
Gaballa MA, Goldman S (2002) Ventricular remodeling in heart failure. J Card Fail 8:S476–S485
Bolognese L, Neskovic AN, Parodi G, Cerisano G, Buonamici P, Santoro GM, Antoniucci D (2002) Left ventricular remodeling after primary coronary angioplasty: patterns of left ventricular dilation and long-term prognostic implications. Circulation 106:2351–2357
Yang F, Liu YH, Yang XP, Xu J, Kaplan A, Carretero OA (2002) Myocardial infarction and cardiac remodelling in mice. Exp Physiol 87:547–555
Zhao ZQ, Corvera JS, Halkos ME, Kerendi F, Wang NP, Guyton RA, Vinten-Johansen J (2003) Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am J Physiol 285:H579–H588
Staat P, Rioufol G, Piot C, Cottin Y, Cung TT, L’Huillier I, Aupetit J-F, Bonnefoy E, Finet G, Andre-Fouet X, Ovize MM (2005) Postconditioning the human heart. Circulation 112:2143–2148
Darling CE, Solari PB, Smith CS, Furman MI, Przyklenk K (2007) ‘Postconditioning’ the human heart: multiple balloon inflations during primary angioplasty may confer cardioprotection. Basic Res Cardiol 102:274–278
Yang X-C, Liu Y, Wang L-F, Cui L, Ge Y-G, Wang H-S, Li W-M, Xu L, Ni Z-H, Liu H-S, Zhang L, Wang T, Jia H-M, Vinten-Johansen J, Zhao Z-Q (2006) Permanent reduction in myocardial infarct size by postconditioning in patients after primary coronary angioplasty. Circulation 114(18):II–812(3795)
Heusch G (2004) Postconditioning: old wine in a new bottle? J Am Coll Cardiol 44:1111–1112
Vinten-Johansen J, Lefer DJ, Nakanishi K, Johnston WE, Brian CA, Cordell AR (1992) Controlled coronary hydrodynamics at the time of reperfusion reduces postischemic injury. Cor Art Dis 3:1081–1093
Sato H, Jordan JE, Zhao Z-Q, Sarvotham SS, Vinten-Johansen J (1997) Gradual reperfusion reduces infarct size and endothelial injury but augments neutrophil accumulation. Ann Thorac Surg 64:1099–1107
Kin H, Zhao ZQ, Sun H-Y, Wang NP, Corvera JS, Halkos ME, Kerendi F, Guyton RA, Vinten-Johansen J (2004) Postconditioning attenuates myocardial ischemia-reperfusion injury by inhibiting events in the early minutes of reperfusion. Cardiovasc Res 62:74–85
Piper HM, Garcia-Dorado D, Ovize M (1998) A fresh look at reperfusion injury. Cardiovasc Res 38:291–300
Piper HM, Schafer AC (2004) The first minutes of reperfusion: a window of opportunity for cardioprotection. Cardiovasc Res 61:365–371
Tsao PS, Aoki N, Lefer DJ, Johnson G, III Lefer AM (1990) Time course of endothelial dysfunction and myocardial injury during myocardial ischemia and reperfusion in the cat. Circulation 82:1402–1412
Kin H, Zatta AJ, Lofye MT, Amerson BS, Halkos ME, Kerendi F, Zhao ZQ, Guyton RA, Headrick JP, Vinten-Johansen J (2005) Postconditioning reduces infarct size via adenosine receptor activation by endogenous adenosine. Cardiovasc Res 67:124–133
Heusch G, Buchert A, Feldhaus S, Schulz R (2006) No loss of cardioprotection by postconditioning in connexin 43-deficient mice. Basic Res Cardiol 101:354–356
Kerendi F, Kin H, Halkos ME, Jiang R, Zatta AJ, Zhao Z-Q, Guyton RA, Vinten-Johansen J (2005) Remote postconditioning: brief renal ischemia and reperfusion applied before coronary artery reperfusion reduces myocardial infarct size via endogenous activation of adenosine receptors. Basic Res Cardiol 100:404–412
Tsang A, Hausenloy DJ, Macanu MM, Yellon DM (2004) Postconditioning-A form of “modified reperfusion” protects the myocardium by activating the P13K-Akt pathway. Circulation 110:III167
Zhu M, Feng J, Lucchinetti E, Fischer G, Xu L, Pedrazzini T, Schaub MC, Zaugg M (2006) Ischemic postconditioning protects remodeled myocardium via the PI3K-PKB/Akt reperfusion injury salvage kinase pathway. Cardiovasc Res 72:152–162
Yang XM, Philipp S, Downey JM, Cohen MV (2005) Postconditioning’s protection is not dependent on circulating blood factors or cells but involves adenosine receptors and requires PI3-kinase and guanylyl cyclase activation. Basic Res Cardiol 100:57–63
Philipp S, Yang X-M, Cui L, Davis AM, Downey JM, Cohen MV (2006) Postconditioning protects rabbit hearts through a protein kinase C-adenosine A2b receptor cascade. Cardiovasc Res 70:308–314
Yang X-M, Proctor JB, Cui L, Krieg T, Downey JM, Cohen MV (2004) Multiple, brief coronary occlusions during early reperfusion protect rabbit hearts by targeting cell signaling pathways. J Am Coll Cardiol 44:1103–1110
Halkos ME, Kerendi F, Corvera JS, Wang NP, Kin H, Payne CS, Sun H-Y, Guyton RA, Vinten-Johansen J, Zhao ZQ (2004) Myocardial protection with postconditioning is not enhanced by ischemic preconditioning. Ann Thorac Surg 78:961–969
Iliodromitis EK, Georgiadis M, Cohen MV, Downey JM, Bofilis E, Kremastinos DT (2006) Protection from postconditioning depends on the number of short ischemic insults in anesthetized pigs. Basic Res Cardiol 101:502–507
Schwartz LM, Lagranha CJ (2006) Ischemic postconditioning during reperfusion activates Akt and ERK without protecting against lethal myocardial ischemia-reperfusion injury in pigs. Am J Physiol 290:H1011–H1018
Wang HC, Zhang HF, Guo WY, Su H, Zhang KR, Li QX, Yan W, Ma XL, Lopez BL, Christopher TA, Gao F (2006) Hypoxic postconditioning enhances the survival and inhibits apoptosis of cardiomyocytes following reoxygenation: role of peroxynitrite formation. Apoptosis 11:1453–1460
Sun H-Y, Wang NP, Halkos M, Kerendi F, Kin H, Guyton RA, Vinten-Johansen J, Zhao ZQ (2006) Postconditioning attenuates cardiomyocyte apoptosis via inhibition of JNK and p38 mitogen-activated protein kinase signaling pathways. Apoptosis 11:1583–1593
Entman ML, Michael L, Rossen RD, Dreyer WJ, Anderson DC, Taylor AA, Smith CW (1991) Inflammation in the course of early myocardial ischemia. FASEB J 5:2529–2537
Vinten-Johansen J, Jiang R, Reeves JG, Mykytenko J, Deneve J, Jobe LJ (2007) Inflammation, proinflammatory mediators and myocardial ischemia-reperfusion injury. Hematol Oncol Clin N Am 21:123–145
Lucchesi BR (2001) Myocardial reperfusion injury-role of free radicals and mediators of inflammation. In: Sperelakis N, Kurachi Y, Terzic A, Cohen MV (eds) Heart Physiology and Pathophysiology, 4th edn. pp 1181–1210
Lefer AM, Tsao PS, Lefer DJ, Ma X-L (1991) Role of endothelial dysfunction in the pathogenesis of reperfusion injury after myocardial ischemia. FASEB J 5:2029–2034
Cohen MV, Yang X-M, Downey JM (2007) The pH hypothesis of postconditioning: Staccato reperfusion reintroduces oxygen and perpetuates myocardial acidosis. Circulation
Jordan JE, Zhao ZQ, Vinten-Johansen J (1999) The role of neutrophils in myocardial ischemia-reperfusion injury. Cardiovasc Res 43:860–878
Kennedy TP, Vinten-Johansen J (2006) A review of the clinical use of anti-inflammatory therapies for reperfusion injury in myocardial infarction and stroke: where do we go from here? Curr Opinion Investigation Drugs 7:229–242
Vinten-Johansen J (2004) Involvement of neutrophils in the pathogenesis of lethal myocardial reperfusion injury. Cardiovasc Res 61:481–497
Meldrum DR (1998) Tumor necrosis factor in the heart. Am J Physiol 274:R577–R595
Lefer AM, Ma X-L, Weyrich A, Lefer DJ (1993) Endothelial dysfunction and neutrophil adherence as critical events in the development of reperfusion injury. Agents Actions 41:127–135
Baxter GF (2002) The neutrophil as a mediator of myocardial ischemia-reperfusion injury: time to move on. Basic Res Cardiol 97:268–275
Olafsson B, Forman MB, Puett DW, Pou A, Cates CU, Friesinger GC, Virmani R (1987) Reduction of reperfusion injury in the canine preparation by intracoronary adenosine: importance of the endothelium and the no-reflow phenomenon. Circulation 76:1135–1145
Pitarys CJ, Virmani R, Vildibill HD Jr, Jackson EK, Forman MB (1991) Reduction of myocardial reperfusion injury by intravenous adenosine administered during the early reperfusion period. Circulation 83:237–247
Jordan JE, Zhao ZQ, Sato H, Taft S, Vinten-Johansen J (1997) Adenosine A2 receptor activation attenuates reperfusion injury by inhibiting neutrophil accumulation, superoxide generation and coronary endothelial adherence. J Pharmacol Exp Ther 280:301–309
Bell RM, Yellon DM (2003) Bradykinin limits infarction when adminstered as an adjunct to reperfusion in mouse heart: the role of P18K, Akt and eNOS. J Mol Cell Cardiol 35:185–193
Yang XM, Krieg T, Cui L, Downey JM, Cohen MV (2004) NECA and bradykinin at reperfusion reduce infarction in rabbit hearts by signaling through PI3K, ERK, and NO. J Mol Cell Cardiol 36:411–421
Peart JN, Gross GJ (2003) Adenosine and opioid receptor-mediated cardioprotection in the rat: evidence for cross-talk between receptors. Am J Physiol 285:H81–H89
Jordan JE, Thourani VH, Auchampach JA, Robinson JA, Wang NP, Vinten-Johansen J (1999) A(3) adenosine receptor activation attenuates neutrophil function and neutrophil-mediated reperfusion injury. Am J Physiol 277:H1895–H1905
Yang Z, Day YJ, Toufektsian MC, Ramos SI, Marshall M, Wang XQ, French BA, Linden J (2005) Infarct-sparing effect of A2A-adenosine receptor activation is due primarily to its action on lymphocytes. Circulation 111:2190–2197
Gross GJ (2003) Role of opioids in acute and delayed preconditioning. J Mol Cell Cardiol 35:709–718
Gross ER, Hsu AK, Gross GJ (2004) Opioid-induced cardioprotection occurs via glycogen synthase kinase β inhibition during reperfusion in intact rat hearts. Circ Res 94:960–966
Chang WL, Lee SS, Su MJ (2005) Attenuation of post-ischemia reperfusion injury by thaliporphine and morphine in rat hearts. J Biomed Sci 12:611–619
Kin H, Zatta AJ, Jiang R, Reeves JG, Mykytenko J, Sorescu G, Zhao Z-Q, Wang N-P, Guyton RA, Vinten-Johansen J (2005) Activation of opioid receptors mediates the infarct size reduction by Postconditioning. J Mol Cell Cardiol 38:827
Hausenloy DJ, Yellon DM (2006) Survival kinases in ischemic preconditioning and postconditioning. Cardiovasc Res 70:240–253
Hausenloy DJ, Tsang A, Yellon DM (2005) The reperfusion injury salvage kinase pathway: a common target for both ischemic preconditioning and postconditioning. TCM 15:69–75
Mykytenko J, Reeves JG, Kin H, Zatta AJ, Jiang R, Guyton RA, Vinten-Johansen J, Zhao Z-Q (2005) Postconditioning reduces infarct size via mitochondrial KATP channel activation during 24 hours of reperfusion. J Mol Cell Cardiol 38:830
Argaud L, Gateau-Roesch O, Raisky O, Loufouat J, Robert D, Ovize M (2005) Post-conditioning inhibits mitochondrial permeability transition. Circulation 111:194–197
Darling CE, Jiang R, Maynard M, Whittaker P, Vinten-Johansen J, Przyklenk K (2005) Postconditioning via stuttering reperfusion limits myocardial infarct size in rabbit hearts: role of ERK1/2. Am J Physiol 289:H1618–H1626
Ytrehus K, Liu Y, Downey J (1994) Preconditioning protects ischemic rabbit heart by protein kinase C activation. Am J Physiol 266:H1145–H1152
Inagaki K, Churchill E, Mochly-Rosen D (2006) Epsilon protein kinase C as a potential therapeutic target for the ischemic heart. Cardiovasc Res 70:222–230
Schulz R, Gres P, Skyschally A, Duschin A, Belosjorow S, Konietzka I, Heusch G (2003) Ischemic preconditioning preserves connexin 43 phosphorylation during sustained ischemia in pig hearts in vivo. FASEB J 17:1355–1357
Inagaki K, Hahn HS, Dorn II GW, Mochly-Rosen D (2003) Additive protection of the ischemic heart ex vivo by combined treatment with ð-Protein kinase C inhibitor and ε-Protein kinase C activator. Circulation 108:869–875
Inagaki K, Chen L, Ikeno F, Lee FH, Imahashi K, Bouley DM, Rezaee M, Yock PG, Murphy E, Mochly-Rosen D (2003) Inhibition of delta-protein kinase C protects against reperfusion injury of the ischemic heart in vivo. Circulation 108:2304–2307
Churchill EN, Szweda LI (2005) Translocation of deltaPKC to mitochondria during cardiac reperfusion enhances superoxide anion production and induces loss in mitochondrial function. Arch Biochem Biophys 439:194–199
Murriel CL, Churchill E, Inagaki K, Szweda LI, Mochly-Rosen D (2004) Protein kinase Cdelta activation induces apoptosis in response to cardiac ischemia and reperfusion damage: a mechanism involving BAD and the mitochondria. J Biol Chem 279:47985–47991
Toma O, Weber NC, Wolter JI, Obal D, Preckel B (2004) Desflurane preconditioning induces time-dependent activation of protein kinase C epsilon and extracellular signal-regulated kinase 1 and 2 in the rat heart in vivo. Anesthesiology 101:1372–1380
Kostyak JC, Hunter JC, Korzick DH (2006) Acute PKCdelta inhibition limits ischaemia-reperfusion injury in the aged rat heart: role of GSK-3beta. Cardiovasc Res 70:325–334
Baines CP, Song C-X, Zheng Y-T, Wang G-W, Zhang J, Wang O-L, Guo Y, Bolli R, Cardwell EM, Ping P (2003) Protein kinase C[epsilon] interacts with and inhibits the permeability transition pore in cardiac mitochondria. Circ Res 92:873–880
Zatta AJ, Kin H, Lee G, Wang N, Jiang R, Lust R, Reeves JG, Mykytenko J, Guyton RA, Zhao ZQ, Vinten-Johansen J (2006) Infarct-sparing effect of myocardial postconditioning is dependent on protein kinase C signalling. Cardiovasc Res 70:315–324
Penna C, Rastaldo R, Mancardi D, Raimondo S, Cappello S, Gattullo D, Losano G, Pagliaro P (2006) Post-conditioning induced cardioprotection requires signaling through a redox-sensitive mechanism, mitochondrial ATP-sensitive K+ channel and protein kinase C activation. Basic Res Cardiol 101:180–189
Hearse DJ, Humphrey SM, Chain EB (1973) Abrupt reoxygenation of the anoxic potassium-arrested perfused rat heart: a study of myocardial enzyme release. J Mol Cell Cardiol 5:395–407
Duilio C, Ambrosio G, Kuppusamy P, DiPaula A, Becker LC, Zweier JL (2001) Neutrophils are primary source of 02 radicals during reperfusion after prolonged myocardial ischemia. Am J Physiol 280:H2649–H2657
Zweier JL, Flaherty JT, Weisfeldt ML (1987) Direct measurement of free radicals generated following reperfusion of ischemic myocardium. Proc Natl Acad Sci 84:1404–1407
Serviddio G, Di Venosa N, Federici A, D’Agostino D, Rollo T, Prigigallo F, Altomare E, Fiore T, Vendemiale G (2005) Brief hypoxia before normoxic reperfusion (postconditioning) protects the heart against ischemia-reperfusion injury by preventing mitochondria peroxyde production and glutathione depletion. FASEB J 19:354–361
Ytrehus K, Myklebust R, Mjos OD (1986) Influence of oxygen radicals generated by xanthine oxidase in the isolated perfused rat heart. Cardiovasc Res 20:597–603
Sun H-Y, Wang NP, Kerendi F, Halkos M, Kin H, Guyton RA, Vinten-Johansen J, Zhao ZQ (2005) Hypoxic postconditioning reduces cardiomyocyte loss by inhibiting ROS generation and intracellular Ca2+ overload. Am J Physiol 288:H1900–H1908
Mykytenko J, Kerendi F, Reeves JG, Kin H, Zatta AJ, Jiang R, Guyton RA, Vinten-Johansen J, Zhao ZQ (2007) Long-term inhibition of myocardial infarction by postconditioning during reperfusion. Basic Res Cardiol 102:90–100
Di Lisa F, Menabo R, Canton M, Barile M, Bernardi P (2001) Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD(+) and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J Biol Chem 276:2571–2575
Griffiths EJ, Halestrap AP (1995) Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem J 307:93–98
Bopassa JC, Ferrera R, Gateau-Roesch O, Couture-Lepetit E, Ovize M (2006) PI 3-kinase regulates the mitochondrial transition pore in controlled reperfusion and postconditioning. Cardiovas Res 69:178–185
Donato M, D’Annunzio V, Saban M, Flor L, Gelpi RJ (2004) Ischemic postconditioning reduced infarct size in normal and hypercholesterolemic rabbit hearts. J Mol Cell Cardiol 37:179
Iliodromitis EK, Zoga A, Vrettou A, Andreadou I, Paraskevaidis IA, Kaklamanis L, Kremastinos DT (2006) The effectiveness of postconditioning and preconditioning on infarct size in hypercholesterolemic and normal anesthetized rabbits. Atherosclerosis 188:356–362
Vinten-Johansen J, Johnston WE, Mills SA, Faust KB, Geisinger KR, DeMasi RJ, Cordell AR (1988) Reperfusion injury after temporary coronary occlusion. J Thorac Cardiovasc Surg 95:960–968
Jolly SR, Kane WJ, Bailie MB, Abrams GD, Lucchesi BR (1984) Canine myocardial reperfusion injury: its reduction by the combined administration of superoxide dismutase and catalase. Circ Res 54:277–285
Bolli R, Becker L, Gross G, Mentzer RM, Balshaw D Jr, Lathrop DA (2004) Myocardial protection at a crossroads: the need for translation into clinical therapy. Circ Res 95:125–134
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Vinten-Johansen, J. Postconditioning: a mechanical maneuver that triggers biological and molecular cardioprotective responses to reperfusion. Heart Fail Rev 12, 235–244 (2007). https://doi.org/10.1007/s10741-007-9024-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-007-9024-3