
Abstract
Lead (Pb), a ubiquitous and potent neurotoxicant, induces several neurophysiological and behavioural changes, while Pb alters the function of multiple organs and systems, it primarily affects the central nervous system. In human adults, encephalopathy resulting from Pb intoxication is often characterized by sleeplessness, poor attention span, vomiting, convulsions and coma; in children, Pb-induced encephalopathy is associated with mental dullness, vomiting, irritability and anorexia; diminished cognitive function resulting in a mental deficit has been also observed during Prolonged exposure to Pb. Pb can produce oxidative stress, disrupt the blood–brain barrier and alter several Ca2+-dependent processes, including physiological processes that involve nitric oxide synthesis on central nervous system in development and adult animals. This review summarizes recent evidence showing that Pb can interfere with the production of nitric oxide and can disrupt the function of nitric oxide synthase. Lead interferes with nitric oxide-related physiological mechanisms, and Pb neurotoxicity may affect processes involved in learning and memory.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lead (Pb) is a ubiquitous pollutant in the ecosystem. It is one of the most useful metals and is detectable in nearly all phases of the inert environment and in all biological systems. Environmental levels of Pb have increased more than 1,000-fold over the past 3 centuries as the result of human activity; the greatest increase occurred between the years 1950 and 2000 (ATSDR 2007). Despite efforts to reduce exposure through regulation, excessive Pb exposure still persists. The general population is primarily exposed to Pb from air and food; in contrast, occupational exposure to Pb occurs in workers employed in Pb refining, welding of lead painted metal, mines, and in battery plants. Over the last few decades, however, Pb emissions in developed countries have decreased markedly due to the introduction of unleaded gasoline (Järup 2003). Blood Pb levels were revised in the general population over the past 3 decades and the results demonstrated the adverse effects of Pb on child neurodevelopment; consequently, the levels of Pb considered hazardous were reduced to 40 μg/dl in 1971, 30 μg/dl in 1975, 10 μg/dl in 1991 (Bellinger and Bellinger 2006) and 1–2 μg/dl in 2004 (EPA 2006); however, Pb is still a significant public health concern.
The biological half-life of Pb may be considerably longer in children than in adults. Pb in blood has an estimated half-life of 35 days, in soft tissue 40 days and in bones 20–30 years (Papanikolaou et al. 2005). Pb has well-characterized effects on each bodily system, Pb toxicity targets are the haematological and cardiovascular systems and the kidneys, bones and teeth (Klein and Snodgrass 2003; ATSDR 2007) but the nervous system is especially susceptible to its effects. Children are particularly sensitive to the effects of the Pb and it is thus considered a primary environmental hazard (Papanikolaou et al. 2005; White et al. 2007); Pb intoxication in young children produces dramatic effects; they may be affected by behavioural disturbances and difficulties in learning and mental concentration, as well as cognitive deficits that include reduced IQ scores even at low levels of exposure. The increased vulnerability to Pb in children is due, in part, to the different exposure pathway and differences in toxicokinetics (Goyer 1993; Goyer and Clarkson 2001; Järup 2003; Bellinger and Bellinger 2006; White et al. 2007). The effects on memory and learning are mainly caused by the action of Pb on several brain structures; however, Pb primarily affects the hippocampus, where Pb neurotoxicity alters molecular mechanisms involving the nitric oxide synthases.
Several reviews have been published about the molecular mechanisms of Pb neurotoxicity, detailing the effect of Pb on different neuronal processes including metal-transporting proteins, ionic channels, glutamatergic synapses, synaptic plasticity, and signalling molecules, among others. The primary purpose of this paper is to describe the mechanisms of Pb neurotoxicity, showing how lead impairs processes involving nitric oxide synthase and how these processes produce serious repercussions on learning and memory.
Neurotoxicity of Pb
Lead is a heavy, low-melting, bluish-gray metal that occurs naturally in the Earth’s crust. In human, about 50 % of Pb is absorbed by inhalation route, while that only 10–15 % is absorbed by oral route; in each case, 90 % of the Pb that enters is retained in the body and distributed to the bones (Links et al. 2001). Once Pb accumulates in other organs, Pb crosses the blood–brain barrier (BBB) and concentrates in the gray matter of the brain (Goyer and Clarkson 2001; Gwalteney-Brant 2002). In humans, acute Pb toxicity is less common that chronic exposure and usually manifests as headache, irritability, abdominal pain and neurological signs; chronic Pb encephalopathy is often characterized by sleeplessness, poor attention span, vomiting, convulsions and coma (Bellinger et al. 1992). In children, Pb encephalopathy is characterized by lethargy, mental dullness, vomiting, irritability and anorexia; in severe cases, prolonged exposure to Pb can decrease cognitive function and increases behaviour disorders, especially aggression and hyperactivity (Bellinger et al. 1992; Gwalteney-Brant 2002; Järup 2003; ATSDR 2007). The neuropathological effects of chronic Pb intoxication include prominence of cerebral and cerebellar capillaries with endothelial cell swelling and necrosis, resulting in severe cerebral oedema caused by enhanced capillary leakage, loss of neuronal cells (Gwalteney-Brant 2002), cytoplasmic vacuolization, hyperchromatic cells, chromatolysis, interstitial oedema (Hirano and Iwata 1989) and demyelination of nerve fibers (Soltaninejad et al. 2003). Ultrastructurally, Pb causes alterations in mitochondria, Golgi apparatus and increment of gliofilaments in astrocytes (Strużyñska et al. 2001). Apoptotic cells with nuclear fragmentation and apoptotic bodies in hippocampal cells have also been described (Sharifi et al. 2002).
The endothelial cells are the first to be exposed to the Pb passage into the brain, and this exposure alters the maturation or differentiation of astrocytes; the endothelial cells show a marked affinity for Pb, resulting in high accumulations especially in mitochondria (Costa et al. 2004). Pb interferes with the phosphorylation of protein kinase C (PKC), which is activated as a consequence of receptor-dependent increases in intracellular Ca2+ concentrations and diacylglycerol (DAG), this activation increase transendothelial permeability (Zengh et al. 2003), leading enters Pb, ions, and water, and producing oedema and brain damage (Bressler and Goldstein 1991). The Pb enters the astrocytes from receptor-operated and voltage-dependent Ca2+ channels that mediate the uptake of Pb ions in these cells; it is recruited and deposited in the lysosome, the nucleus and other organelles of astroglia, a process that presumably occurs via Pb-binding proteins (Tiffany-Castiglioni and Qian 2001), astrocytes thus play a critical role in the brain, protecting neurons against Pb toxicity (Harry et al. 1996). A major intermediate filament protein found in astrocytes, glial protein fibrillary acidic protein (GFAP), has been suggested as an early indicator of neurotoxic insult (O’Callaghan and Jensen 1992), commonly, exposure of astrocytes to neurotoxic insults results in astrogliosis, which increases the synthesis and concentration of GFAP (Van Den Berg et al. 1996; Strużyñska et al. 2001). Several studies in Pb-exposed rats have shown that the expression, synthesis, and concentration of GFAP, as well as the number of astrocytes, were increased in specific brain regions (Fig. 1), this increase is associated with the formation of reactive gliosis in the rat brain, suggesting a primary response of astrocytes to Pb (Selvin-Testa et al. 1997; Stoltenburg-Didinger et al. 1996; Strużyñska et al. 2001; Tiffany-Castiglioni 1993; Tiffany-Castiglioni and Qian 2001; Harry et al. 1996; Strużyñska et al. 2001).

Cells exposed to Pb can alter the modulation of cell signals through different mechanisms such as: a interfering with processes regulate by Ca2+ that subsequently change transcription factors expression and/or activity; b affecting the LPO leading to oxidative stress altered; c interfering with enzymatic function; d induced alterations of the major nueurotransmitter systems and e interfering with protein components and/or activity. PKC Protein Kinase C, GFAP Glial Protein Fibrillary Acidic, NOS Nitric Oxide Synthase, GPx Glutathione Peroxidase, SOD Super Oxide Dismutase, CAT Catalase, Ca 2+ Calcium, LPO Lipid Peroxidation
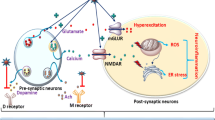
As previously mentioned, Pb can affect the nervous system through multiple pathways. Pb acts by mimicking Ca2+ action and/or disrupting Ca2+ homeostasis. Pb2+ is a divalent cation, and its neurotoxic action might be related to its ability to substitute for Ca2+ and, therefore, inhibit or increase the action of Ca2+ (Audesirk 1993). The interaction between Pb2+ and Ca2+ suggests that Pb2+ may gain entry into cells through one or more of the different types Ca2+ channels expressed in various cells (Bridges and Zalups 2005; ATSDR 2007). Likewise several neurotransmitters are also affected by Pb interaction, in animals exposed to low doses of Pb during development, high levels of dopamine and enhanced catecholaminergic neurotransmission have been observed in the cerebral cortex, hippocampus and cerebellum (Leret et al. 2002; Devi et al. 2005). Rats exposed to high concentrations of Pb have a decrease in norepinephrine, epinephrine and dopamine levels in the cerebral cortex, hippocampus and cerebellum, demonstrating that Pb exposure affects the neurotransmitter system in the developing rat brain (Fig. 1) (Devi et al. 2005). Glutamate is the major excitatory neurotransmitter in the brain ionotropic and metabotropic receptors mediate the action of glutamate via activation of the N-methyl-D-aspartate receptor (NMDAr) and play a central role in learning and memory. The NMDAr, which is mediated by Ca2+, activates protein kinase A (PKA), mitogen-activated protein kinase (MAPK) and calcium/calmodulin-dependent protein kinase (CAMK) pathways, which converge at the cyclic-AMP-response element-binding protein (CREB) (Fig. 2) (Toscano and Guilarte 2005).

In neurons, NO synthesis is initiated by a glutamate release leads Ca2+-influx into the cell, and binds to calmodulin. The binding of Ca2+/calmodulin complex to the catalytic site of NOS induce the oxidation of l-arginine to synthetize citruline and NO. Pb also affects the processes for NOS expression. It is possible that the Pb2+ is negatively acting on Ca2+-dependent transcription elements for CREB and AP-1 for nNOS and eNOS respectivelly. NO nitric oxide, Ca 2+ Calcium, Ca 2+ /CaM Calcium-calmodulin, NOS Nitric Oxide Synthase structure, CREB cAMP response element binding protein, AP-1 activator protein 1
The two pathways that have received more attention are calmodulin and PKC. Ca2+ induces a conformational change in calmodulin that converts the protein to its active form. Pb2+ acts by displacing Ca2+ ions bound to calmodulin; the activation of calmodulin by Pb2+ results in protein phosphorylation altering cAMP messenger pathways (Goyer 1997; Goyer and Clarkson 2001). Normally, Ca2+ is responsible for activating PKC, which is a serine/threonine protein kinase involved in many processes such as synaptic transmission, neurotransmitter synthesis, dendritic branching, as well as the transduction of signals generated by a wide variety of effectors through specific receptors (Bressler et al. 1999). Pb is better activator of PKC than Ca2+ (Goyer 1997), and the activation of PKC induce genes that regulate the formation of the AP-1 transcriptional regulatory complex (Bressler et al. 1999), this activation disturbs signalling mechanisms in the hippocampus, causing impairment on long-term potentiation and memory in adult rats (Fig. 2), (Altmann et al. 1993; Gu et al. 2005).
Pb and oxidative stress
Oxidative damage is considered an important factor in Pb neurotoxicity, experimental evidence suggests that Pb induces oxidative stress and exerts some of its toxic effects through the disruption of pro-oxidant/anti-oxidant balance, which can lead to brain injury via oxidative damage (Oteiza et al. 1995; Adonaylo and Oteiza 1999; Antonio et al. 2003; Daniel et al. 2004; Villeda-Hernández et al. 2001; Villeda-Hernández et al. 2006). Pb exposure might induce changes in the activities of antioxidant enzymes that can be primarily attributed to the high affinity of Pb for sulphhydryl groups or metal cofactors in antioxidant enzymes (Gurer and Ercal 2000). Several authors have reported that Pb can induce significative decrease in the activity of superoxide dismutase (SOD) and catalase (CAT) in adult mouse and rat brains exposed to Pb (Skoczynska et al. 1993; Nehru and Kanwar 2004; Moreira et al. 2001a). Similar results were obtained by Wang et al. (2006), who found that the activity of SOD, Glutathione peroxidase (GPx) and reduced glutathione (GSH) were decreased significantly in 21-day-old rat brains exposed to Pb during pregnancy. However, contrary reports have indicated a gradual increase in the activity of antioxidant enzymes such as GPx, glutathione-reductase, CAT and SOD, mainly in the cerebellum and hippocampus (Antonio et al. 2003; Bennet et al. 2007). These studies support that Pb enhances lipoperoxidation (LPO) induced by H2O2 and iron and increases fluxes of O−2 and H2O2 (Gurer et al. 1988) resulting from a region-specific oxidative stress response in the brain (Bennet et al. 2007). It is unclear whether oxidative stress is the cause or the consequence of the toxic effect of Pb.
Nitric oxide (NO)
Nitric oxide is a signalling molecule with a variety of physiological functions. It is an unstable molecule with a molecular weight of 30 kDa and a short half-life of approximately 5–30 s. NO is a lipophilic gas that passes through the cellular membrane easily and can reach a diffusion distance of 150–300 μm in 4–15 s. It is produced by the stoichiometric conversion of l-arginine to l-citrulline via different isoforms of nitric oxide synthases (NOS) (Moncada et al. 1989; Kröncke et al. 1997; Bruckdorfer 2005). Molecular cloning experiments have led to the identification of three isoforms of NOS: neuronal NOS (nNOS or NOS-I), endothelial NOS (eNOS or NOS-III) and inducible NOS (iNOS or NOS-II) (Doyle and Slater 1997). NOS is widely distributed; it has been localized and studied in the cerebellum, hippocampus, cerebral cortex, corpus striatum, thalamus, amygdala, and olfactory bulb, among other regions of the central nervous system (Dawson and Dawson 1996; García-Arenas et al. 1999).
Nitric oxide is considered a free radical because it has an unpaired electron. NO reacts rapidly with O−2· to produce the peroxynitrite anion (ONOO−), which protonates at relevant pH to form peroxynitrous acid (ONOOH). Both ONOO− and ONOOH are potent oxidizers; ONOOH exhibits hydroxyl radical (·OH)—like activity, which can initiate a chain reaction generating numerous toxic metabolites (Aschner 1996). Nitrosative stress may lead to nitrosylation reactions that can alter the structure of proteins and so inhibit their normal function. In pathological conditions, NO is synthesized in excess and interacts with superoxide (·O2 −) to give rise to reactive oxygen and nitrogen species (ROS and RNS respectively) cascades (Valko et al. 2006).
Nitric oxide plays an important role in the function of several peripheral organs, including the digestive, respiratory and urogenital tracts (Toda and Okamura 2003). Likewise, is associated with brain processes that involve synaptogenesis (Estrada and Murillo-Carretero 2005), cerebral blood flow (Santizo et al. 2000), neuroendocrine secretion and neurotransmission (Chrousos 1995). NO is implicated in the regulation of the neurotransmitter release pathway that inhibits respiratory complexes contributing to glutamate excitotoxicity or stimulation by activation of guanylyl cyclase, which augment the phosphorylation of synaptic vesicles (Knott and Bossy 2009), is important its participation in immune responses involving cytokines and endotoxins, by produce high NO concentrations with cytotoxic effects on target cells (Simonian and Coyle 1996; Kröncke et al. 1997; Sun et al. 2005; Garthwaite 2008a, b). NO play an important role in Long Term Potentiation (LTP), LTP is a form of synaptic plasticity that is believed to form the cellular basis for learning and memory. It is well know that activation the NMDAr in brain are important in LTP induction, because the overstimulation of NMDAr by glutamate leads to Ca2+ overload in the cell and consequently activation of Ca2+ sensitive enzymes such as cNOS that induces NO biosynthesis (Zhang et al. 1998; García-Arenas et al. 2004; Hawkins et al. 1998).
Likewise, recent findings shown an association between increased concentrations of NO and diseases such as: migraine and epilepsy where the NO can exert inhibitory and excitatory effects on GABA-ergic transmission (Talarek and Fidecka 2003). Studies in humans and rodents shown that exist an increase of NO production in Parkinsons’s Disease, which is associated with a progressive loss of dopaminergic neurons in substance nigra (Burns et al. 1983; Kühn et al. 2003); Alzheimer disease show an increase on the three isoforms of NOS indicating an important role for NO in the pathomechanism of disease (Thorns et al. 1998; Lüth et al. 2001). Several pathological processes how ischemia, hypoxia, stroke, infectious agents, tumours and autoimmune diseases (Zhou and Zhu 2009) can involve NO alterations.
Nitric oxide synthase (NOS)
Neuronal nitric oxide synthase (nNOS)
Neuronal nitric oxide synthase is an enzyme that consists of 1,434 amino acids and has a molecular weight of 160.8 kDa. It is localized in human chromosome number 12 (Boissel and Schwarz 1998; Bruckdorfer 2005). NO synthesis is initiated by the presynaptic release of glutamate, which binds to NMDA, AMPA/Kainate and metabotropic receptors and allows Ca2+ to enter into the cell. The binding of Ca2+/calmodulin to nNOS induces the conformational change of the dimer and allows electrons to flow from the reductase segment of the enzyme, which contains the flavoprotein- (FAD- and FMN-) binding domains, to the catalytic site. This electron flow facilitates the oxidation of l-arginine and the synthesis of l-citrulline and NO (Alderton et al. 2001; Bruckdorfer 2005; Zhou and Zhu 2009). NO quickly spreads in the retrograde direction to the presynaptic neuron, where it united to guanylate cyclase activates the synthesis of cGMP or modifies the release of neurotransmitters such as acetylcholine, aspartate and glutamate (Fig. 2). NO can also return to the site of its original synthesis and influence the release of neurotransmitters in adjacent synapses (Talavera et al. 2003; García and Baltrons 2004; Bruckdorfer 2005). NO is released after stimulation and diffuses from the site of its production (either a neuronal or glial cell) to affect neurons up to 10-400 μm away (Talavera et al. 2003). nNOS is localized in subsets of neurons and astrocytes belonging to different anatomical and functional regions (Dawson and Dawson 1996). Although nNOS-derived NO is a critical molecule in mediating synaptic plasticity and neuronal signalling, it change from a physiological neuromodulator to a neurotoxic factor when an excessive amount of NO is produced (Zhou and Zhu 2009).
Endothelial nitric oxide synthase (eNOS)
The eNOS isoform is constitutively expressed in endothelial cells and cardiac myocytes; it has 1,203 amino acids and a molecular weight of 133 kDa, and it is located in human chromosome number 7 (Chatterjee and Catravas 2008). Although also has been found in astrocytes (Wiencken and Casagrande 1999) and hippocampal neurons (Dinerman et al. 1994); have a similar structure to nNOS, contains an amino terminal that differs from other NOS isoforms due to is especially proline-rich and has a consensus target sequence for myristoylation via acyltransferases. eNOS is a Ca2+-dependent enzyme (Marsden et al. 1993; Mac-Micking et al. 1997).
Inducible nitric oxide synthase (iNOS)
Inducible NOS (iNOS) is regulated predominantly at the transcriptional level and is likely involved in immune reactions. It has a molecular weight of 131 kDa, is composed of 1,153 amino acids and is located in human chromosome number 17 (Alderton et al. 2001). This enzyme is Ca2+ independent because calmodulin remains tightly bound to the protein; after induction, iNOS continuously produces NO until the enzyme is degraded (Mac-Micking et al. 1997). The iNOS isoform has a structure similar to nNOS and eNOS; it is composed of one oxygenase domain and one reductase domain with a catalytic site, it contains binding sites for FAD, FMN and NADPH (Bruckdorfer 2005). The iNOS isoform is not expressed in the healthy brain but is found primarily in immune cells or glial cells (astrocytes and microglia), as it is activated in response to pathogen recognition and cytokine release (Simonian and Coyle 1996; Kröncke et al. 1997).
Regulation and expression of NOS
The Ca2+ then binds to calmodulin to form the Ca2+/calmodulin complex that, in turn, activates Ca2+-responsive signalling pathways that converge on specific transcription factor such as CREB (Lonze and Ginty 2002); this pathways are PKA, CAMK and MAPK, activate CREB by phosphorylation at serine-133; CREB belongs to the bZip superfamily of transcription factors that contain a C-terminal basic domain that mediates DNA binding, and a leucine zipper domain that facilitates dimerization (Toscano and Guilarte 2005; Riccio et al. 2006).
The structure of nNOS gene is composed by 29 exons and its expression is regulated by membrane depolarization, as well the subsequent interaction of CREB and/or CREB-related family members with the calcium response elements (CRE) sites of the nNOS, which is contained on the promoter region within exon 2 immediately upstream of the transcription start site (Sasaki et al. 2000). Many of these processes are calcium-dependent and due to nNOS catalytic activity is also Ca2+-dependent, there is a unique potential for both simultaneous transcriptional and catalytic activity-mediated regulation of nNOS/NO (Wang et al. 1999). Regulation of nNOS transcription may be controlled by several alternatives of splicing of exon 1 to exon 2; the importance of exon 2 is mainly due to able regulating nNOS expression levels in several tissues and different physiological conditions (Boissel and Schwarz 1998; Dawson et al. 1998). The regulation of the expression of nNOS is a Ca2+-regulated gene through the interactions with CREB and these interactions are involved on diverse pathological and physiological processes such as learning and memory (Lonze and Ginty 2002; Toscano and Guilarte 2005).
The eNOS is encoded by 26 exons spanning 21–22 kb of genomic DNA, the promoter region of eNOS contains numerous binding sites for transcription factor such as AP-1, AP-2, NF-IL6 between others, as well as sites of the estrogen- and glucocorticoid-responsive element (Li et al. 2002). The expression of eNOS and posttranscriptional regulation gene occurs in the form of alternative mRNA splicing, and can be altered by different compounds (TNFα, thrombin, amphotericin B, glucocorticoids) or pathophysiological conditions (hypercholesterolemia, artherosclerosis, diabetes, hypertension) (Li et al. 2002).
The iNOS gene contains 27 exons showing a sequence of 16 kb, in cloned iNOS promoter that exhibits homologies to binding sites for numerous transcription factors involved, (AP-1, CAR, NRF, Nuclear Factor-κB lipopolisacharides/cytokines) that mediate induction and regulation of promoter activity (Akar and Feinstein 2009). The regulation of iNOS expression by transcriptional and posttranscriptional mechanisms is the main regulatory step to control iNOS activity. Usually, iNOS synthesizes NO continuously until the enzyme is degraded; relatively little information exist on iNOS splice variants (Pautz et al. 2010).
Effect of Pb on brain NOS
The primary mechanism of Pb toxicity is related to its ability to establish interactions with coordinated divalent cations such as Ca2+ and Zn. Pb can alter the activity and expression of nNOS and eNOS in different brain regions primarily because Pb can mimic Ca2+ at binding sites, this binding may prevent the accessibility of Ca2+ to NOS, thus leading to the decreased activity of nNOS/eNOS and resulting in reduced NO production in different brain regions (Selvin-Testa et al. 1997). Several studies have reported a decrease in nNOS activity in animals exposed to Pb, Chen et al. (2000) founded that rats exposed to different concentrations of Pb displayed a decrease in NO production as indicated by decreased nitrite and nitrate levels. Others studies have also shown that NOS activity in the hippocampus, cerebral cortex and cerebellum is inhibited significantly by Pb exposure; the degree of this inhibitory effect depends on the time span of exposure and the concentration of Pb; however, this accumulation does not necessarily account for the inhibition of NOS calcium-dependent activity in specific regions (Zhu et al. 2005). García-Arenas et al. (1999) measured the activity of calcium-dependent NOS and iNOS in both the synaptosomes and capillaries of mice exposed to Pb and founded that NOS was inhibited in synaptosomal fractions throughout the brain. In another study, the same authors founded a specific dose-dependent decrease of NOS activity in the hippocampus and cerebellum, but not in the cortex or brain stem, of adult rats exposed to Pb (García-Arenas et al. 2004); It also seems that the presence of high Pb concentrations enhances iNOS activity (García-Arenas et al. 1999).
Pb also affects the processes involved in NOS expression. It is possible that the cation negatively acts on Ca2+-dependent transcription elements for nNOS (CREB, cAMP) and eNOS (AP-1, activator protein 1) to decrease protein expression (Fig. 2) (Lonze and Ginty 2002; Riccio et al. 2006; Sasaki et al. 2000; Wang et al. 1999; Dawson et al. 1998; Li et al. 2002). Immunohistochemical studies have shown changes in the pattern of NOS expression in the cortex and hippocampus of adult and developing rats exposed to Pb (Selvin-Testa et al. 1997; Chung et al. 2004). More recent reports have confirmed these results; Nava-Ruíz et al. (2010) showed that the expression patterns of nNOS and eNOS were diminished in the hippocampus of adult rats exposed to Pb using a sub-acute model. In addition, in vitro studies have shown that under the stated experimental conditions, Pb-acetate may decrease the expression of the iNOS protein in C6 glia cells; (Tiffany-Castiglioni et al. 1990).
Effect of Pb on learning and memory
Learning is defined as the processes of acquiring new information or skills, whereas memory refers to the persistence of learning that can be revealed/recalled at a later time. Memory is the usual consequence of learning and reflects enduring changes in the nervous system that result from transient experiences. The hippocampus is an anatomical structure responsible for diverse memory processes that include the spatial and temporal separation of events; this separation is associated with several types of learning and memory formation through long-term potentiation (LTP). LTP results from an increase in the strength of synaptic transmission, which can last from hours to days. Moreover, the LTP found in the hippocampus has been detected in other brain areas such as the amygdala and cortex, as well as their related limbic structures in the mammalian brain (Squire and Knowlton 1995). The CA1 region of the hippocampus has shown both NOS activity and LTP, a type of synaptic plasticity in the hippocampus that is believed to contribute to declarative forms of learning such as spatial learning (Xu et al. 1998; White et al. 2007).
Experimental studies indicate that Pb is accumulated in several brain regions and is highly concentrated in the hippocampus (Villeda-Hernández et al. 2001; García-Arenas et al. 2004; Nava-Ruíz et al. 2010). The possible deleterious effect of Pb might be related to its interference with Ca2+-dependent cellular processes, an interference that significantly affects the induction of LTP that is mediated by NO regulation. This effect on LTP would explain why learning and memory are affected by chronic Pb exposure (Chetty et al. 2000). Other authors have reported changes in LTP induction in learning and memory pathways in developing and adult animals exposed to Pb; Carpenter et al. (1994) founded that Pb can block LTP at micromolar concentrations in rats exposed to Pb. Similar results were reported by Gilbert and Mack (1990) and Robinson and Reed (1992), who both reported that LTP in the CA1 area of the hippocampus and in the dentate gyrus can be readily blocked by NMDAr antagonists (Fig. 2). García-Arenas et al. (2004) showed that LTP induction was affected in a dose-dependent manner in adult rats exposed to 250 and 500 ppm of acetate of Pb; likewise, it has been reported that Pb alters the amplitude of LTP in hippocampal slices, demonstrating that low-level lead exposure can reduce structural plasticity in adults via neurogenesis (Zhao et al. 1999). Moreover, Altmann et al. (1993) founded a decrease in population spike activity in the hippocampal CA1 region; similarly, other studies have observed an increase in the sensitivity threshold of the LTP blocking response of NMDAr in Pb-exposed rodent hippocampus, indicating that Pb induces a decrease in the magnitude and retention time of synaptic plasticity (Gilbert et al. 1996; White et al. 2007).
Weiss et al. (1998) also founded a neuronal phenotype that was particularly vulnerable to Pb in the hippocampus of rats exposed to Pb during the development, this phenotype was positive for nNOS and displayed Pb-induced changes in NMDAr expression. This discovery indicates that Pb can affect the production of NO, which is a messenger essential for hippocampal LTP (Toscano and Guilarte 2005).
Several studies of cognitive deficits related with NOS activity and NO production have been documented in rats and nonhuman primates models after exposition to Pb (O’Dell et al. 1991; Jett et al. 1997; García-Arenas et al. 2004); when the hippocampal NO levels of mice exposed to Pb were decreased, the mice displayed a reduced ability to learn and memorise on a water T-maze test, suggesting the presence of Pb-induced deficits in learning and memory processes (Sun et al. 2005). Kuhlmann et al. (1997) showed that rats exposed to Pb during different developmental periods were tested as adults in a water maze test, presenting long-term changes in cognitive functions. Likewise, recent results have shown that Pb exposure during gestation can cause pups hyperactivity, decrease exploratory behaviour and prolonged learning/memory deficits in young adult rats (Moreira et al. 2001b; Yang et al. 2003; Song et al. 2006). Experiments performed in monkeys treated with Pb revealed deficits in function a variety of behavioural task, showing to display learning and/or memory impairment (Rice 1993, 1996). In summary these studies shown that in animal models the cognitive functions are altered to Pb exposure at different doses, for different time periods affecting the nNOS activity and reflecting a decrease in the production of NO, mainly in hippocampus producing a cognitive (learning and memory) disrupt, likewise these dates are agreement with epidemiological studies related in humans that support the association between blood Pb levels and intellectual impairment in children with deficit and cognitive development impairments (Canfield et al. 2003; Al-Saleh et al. 2001; Bellinger and Needleman 2003; Mendola et al. 2002; Emory et al. 2003; Gomaa et al. 2002).
Final commentary
Pb is a neurotoxic metal that causes health problems in developed and developing countries. The central nervous system is especially sensitive to Pb, displaying functional alterations mainly during development, in humans and experimental animals. These alterations are due to time and pathways exposure as well as the differences toxicokinetics of Pb. In this review, we summarized the findings related to Pb-induced neuronal damage and the possible mechanism involved in NO synthesis mediating activation and/or inhibition of NOS generating disruption on the behaviour and their effect over NOS gene expression. These results indicate that impaired NO production in animals exposed to Pb can affect memory and learning processes. Future investigations must address those neuronal mechanisms in detail in order to understand Pb-induced damage over NOS function, responsible of NO synthesis.
References
Adonaylo VN, Oteiza PI (1999) Lead intoxication: antioxidant defences and oxidative damage in rat brain. Toxicology 135:77–85
Agency for Toxic Substance and Disease Registry (ATSDR) (2007) Toxicological profile for lead. U.S. Department of Health and Humans Services, Public Health Service, Centres for Diseases Control, Atlanta, GA
Akar CA, Feinstein DL (2009) Modulation of inducible nitric oxide synthase expression by sumoylation. J Neuroinflammation 6:12–21
Alderton WK, Cooper CE, Knowles RG (2001) Nitric oxide synthases: structure, function and inhibition. Biochem J 357:593–615
Al-Saleh I, Nester M, DeVol E et al (2001) Relationship between blood lead concentrations, intelligence, and academia achievement of Arabian schoolgirls. Int J Hyg Environ Health 204:165–174
Altmann L, Weinsber F, Sveinsson K et al (1993) Impairment of long-term potentiation and learning following chronic lead exposure. Toxicol Lett 66:105–112
Antonio MT, Corredor L, Leret ML (2003) Study of the activity of several brain enzymes like markers of the neurotoxicity induced by perinatal exposure to lead and/or cadmium. Toxicol Lett 143:331–340
Aschner M (1996) The functional significance of brain metallothioneins. FASEB J 10:1129–1136
Audesirk G (1993) Electrophysiology of lead intoxication: effects on voltage-sensitive ion channels. Neurotoxicology 14:137–148
Bellinger CD, Bellinger MA (2006) Childhood lead poisoning: the torturous path from science to policy. J Clin Invest 116:853–957
Bellinger DC, Needleman HL (2003) Intelectual impairment and blood lead levels. N Engl J Med 349:500–502
Bellinger DC, Stiles KM, Needelman HL (1992) Low-level lead exposure, intelligence and academic achievement: a long-term follow-up study. Pediatrics 90:855–861
Bennet C, Rajanna B, Sharada R et al (2007) Region specific increase in the antioxidant enzymes and lipid peroxidation products in the brain of rats exposed to lead. Free Radic Res 41:267–273
Boissel JP, Schwarz PM (1998) Neuronal-type NO synthase: transcript diversity and expressional regulation. Nitric Oxide 2:337–349
Bressler JP, Goldstein GW (1991) Mechanisms of lead neurotoxicity. Biochem Pharmacol 41:479–484
Bressler J, Kim KA, Chakraborti T et al (1999) Molecular mechanisms of lead neurotoxicity. Neurochem Res 24:595–600
Bridges CC, Zalups RK (2005) Molecular and ionic mimicry and the transport of toxic metals. Toxicol Appl Pharmacol 204:274–308
Bruckdorfer R (2005) The basics about nitric oxide. Mol Aspects Med 26:3–31
Burns RS, Chiueh CC, Markey SP et al (1983) A primate model of parkinsonism: selective destruction of dopaminergic neurons in the pars compacta of the substantia nigra by N-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine. Proc Natl Acad Sci USA 80:4546–4550
Canfield RL, Kreher DA, Cornwell C et al (2003) Low level lead exposure executive functioning, and learning in early childhood. Child Neuropsychol 9:35–53
Carpenter DO, Matthews MR, Parsons PJ et al (1994) Long-term potentiation in the piriform cortex is blocked by lead. Cell Mol Neurobiol 14:723–733
Chatterjee A, Catravas JD (2008) Endothelial nitric oxide (NO) and its pathophysiologic regulation. Vasc Pharmacol 49:134–140
Chen MS, Huber AB, Vander Daar ME et al (2000) Nogo A is a myelin-associated neurite outgrowth inhibitor and an antigen for monoclonal antibody IN-1. Nature 403:434–439
Chetty CS, Reddy GR, Murthy KS et al (2000) Perinatal lead exposure alters the expression of neuronal nitric oxide synthase in rat brain. Int J Toxicol 20:113–120
Chrousos GP (1995) The hypothalamic-pituitary-adrenal axis and immune-mediated inflammation. N Engl J Med 332:1351–1362
Chung YE, Kim YS, Lee WB (2004) Distribution of neuronal nitric oxide synthase-immunoreactive neurons in the cerebral cortex and hippocampus during postnatal development. J Mol Histol 35:765–770
Costa LG, Aschner M, Vitalone A et al (2004) Developmental neuropathology of environmental agents. Annu Rev Pharmacol 44:87–110
Daniel S, Limson JL, Dairam A (2004) Through metal binding, curcumin protects against lead-and cadmium-induced lipid peroxidation in rat brain homogenates and against lead-induced tissue damage in rat brain. J Inorg Biochem 98:266–275
Dawson VL, Dawson TM (1996) Nitric oxide actions in neurochemistry. Neurochem Int 29:97–110
Dawson TM, Sasaki M, Gonzalez-Zuluta M et al (1998) Regulation of neuronal nitric oxide synthase and identification of novel nitric oxide signaling pathways. Prog Brain Res 118:3–11
Devi CB, Reddy GH, Prasanthi RP et al (2005) Developmental lead exposure alters mitochondrial monoamine oxidase and synaptosomal chatecolamine levels in rat brain. Int J Dev Neurosci 23:375–381
Dinerman JL, Dawson TM, Schell J, Snowman A, Syner SH (1994) Endothelial nitric oxide synthase localized to hippocampal pyramidal cells: implications for synaptic plasticity. PNAS 91:4214–4218
Doyle CA, Slater P (1997) Localization of neuronal and endothelial nitric oxide synthase isoforms in human hippocampus. Neuroscience 76:387–395
Emory E, Ansari Z, Patillo R et al (2003) Maternal blood lead effects on infant intelligence at age 7 months. Am J Obstet Gynecol 188:S26–S32
Environmental Protection Agency (2006) U.S., Air quality criteria for lead Volume I and II of II, Research Triangle Park NC: National Center for Environmental Assessment-RTO Office
Estrada C, Murillo-Carretero M (2005) Nitric oxide and neurogenesis in health and disease. Neuroscientist 11:294–307
García A, Baltrons MA (2004) The nitric oxide/cyclic GMP pathway in CNS glial cells. Adv Mol Cell Biol 31:575–593
García-Arenas G, Claudio L, Pérez-Severiano F et al (1999) Lead acetate exposure inhibits nitric oxide synthase activity in capillary and synaptosomal fractions of mouse brain. Tox Sci 50:244–248
García-Arenas G, Ramírez-Amaya V, Balderas I et al (2004) Cognitive deficits in adult rats by lead intoxiation are related with regional specific inhibition of cNOS. Behav Brain Res 149:49–59
Garthwaite J (2008a) Glutamate, nitric oxide and cell signaling in nervous system. Trends Neurosci 14:60–67
Garthwaite J (2008b) Concepts of neural nitric oxide-mediated transmission. Eur J Neurosci 27:2783–2802
Gilbert ME, Mack CM (1990) The NMDA antagonist, MK-801, suppresses long-term potentiation, kindling, and kindling-induced potentiation in the perforant path of the unanesthetized rat. Brain Res 519:89–96
Gilbert ME, Mack CM, Lasley SM (1996) Chronic developmental lead (Pb2+) exposure increases threshold for long-term potentiation in the rat dentate gyrus in vivo. Brain Res 736:125–134
Gomaa A, Howard H, Bellinger D (2002) Maternal bone lead as an independent risk factor for fetal neurotoxicity: a perspective study. Pediatrics 110:110–118
Goyer RA (1993) Lead toxicity: current concerns. Environ Health Perspect 100:177–187
Goyer RA (1997) Toxic and essential metals interactions. Annu Rev Nutr 17:37–50
Goyer RA, Clarkson TW (2001) Toxic effects of metals. In: Klaassen CD (ed) Casarett and Doull’s, toxicology: the basic science of poisons, 6th edn. McGraw-Hill, New York, pp 811–867
Gu Y, Wang L, Xiao C et al (2005) Effects of lead on voltaje-gated sodium channels in rat hippocampal CA1 neurons. Neuroscience 133:679–690
Gurer H, Ercal N (2000) Can antioxidants be beneficial in the treatment of lead poisoning? Free Radic Biol Med 29:927–945
Gurer H, Ozgunes H, Neal R et al (1988) Antioxidant effects of N-acetylcysteine and succiner in red blood cells from lead-exposed. Toxicology 128:181–189
Gwalteney-Brant SM (2002) Heavy metals. In: Haschek WM, Rosseaux CG, Wallig AM (eds) Handbook of toxicologic pathology, 2nd edn. Academic Press, New York, pp 701–732
Harry JG, Schmitt JT, Gong Z et al (1996) Lead-induced alterations of glial fibrillary acidic protein (GFAP) in the developing rat brain. Toxicol Appl Pharmacol 139:84–93
Hawkins RD, Son H, Arancio O (1998) Nitric oxide as a retrograde messenger during long-term potentiation in hippocampus. Prog Brain Res 72:155–172
Hirano A, Iwata M (1989) Neuropathology of lead intoxication. In: Vinken PJ, Beuyn GW (eds) Intoxication of the nervous system. North-Holland Publishing Company, New York, pp 35–64
Järup L (2003) Hazards of heavy metal contamination. Br Med Bull 68:67–182
Jett DA, Kuhlmann AC, Farmer SJ et al (1997) Age-dependent effects of developmental lead exposure on performance in the Morris water maze. Pharmacol Biochem Behav 57:271–279
Klein GL, Snodgrass WR (2003) Heavy metal toxicology. In: Caballero B, Trugo L, Finglas P (eds) Encyclopedia of food sciences and nutrition, 2nd edn. Elsevier, Amsterdam, pp 3050–3057
Knott AB, Bossy WE (2009) Nitric oxide in health and disease of the nervous system. Antioxid Redox Signal 11:541–553
Kröncke DK, Fehsel K, Kolb BV (1997) Nitric oxide: cytotoxicity versus cytoprotection-how, why, when and where. Nitric Oxide 1:107–120
Kuhlmann AC, McGlothan JL, Guilarte TR (1997) Developmental lead exposure causes spatial learning deficits in adult rats. Neurosci Lett 233:101–104
Kühn K, Wellen J, Link N et al (2003) The mouse MPTP model: gene expression changes in dopaminergic neurons. Eur J Neurosci 17:1–12
Leret ML, García-Uceda F, Antonio MT (2002) Effects of maternal lead administration on momoaminergic, GABAergic and glutamatergic systems. Brain Res Bull 58:469–473
Li H, Wallareth T, Münzel T et al (2002) Regulation of endothelial-type NO synthase expression in pathophysiology and in response to drugs. Nitric Oxide 7:149–164
Links JM, Schwartz BS, Simon D et al (2001) Characterization of toxicokinetics and toxicodynamics with linear systems theory: application to lead-associated cognitive decline. Environ Health Perspect 9:361–368
Lonze BE, Ginty DD (2002) Function and regulation of CREB family transcription factors in the nervous system. Neuron 35:605–623
Lüth HJ, Holzer M, Gärtner U et al (2001) Expression of endothelial and inducible NOS-isoforms is increased in Alzheimer’s disease, in APP23 transgenic mice and after experimental brain lesion in rat: evidence for an induction by amyloid pathology. Brain Res 913:57–67
Mac-Micking J, Xie QW, Nathan C (1997) Nitric oxide and macrophages function. Annu Rev Immunol 15:323–350
Marsden PA, Heng HHQ, Scherer SW et al (1993) Structure and chromosomal localization of the human constitutive endothelial nitric oxide synthase gene. J Biol Chem 268:17478–17488
Mendola P, Selevan SG, Gutter S et al (2002) Environmental factors associated with the spectrum of neurodevelopmental deficits. Ment Retard Dev Disabil Res Rev 8:188–197
Moncada S, Palmer RMJ, Higgs EA (1989) Biosynthesis of nitric oxide from l-arginine: a pathway for regulation of cell function and communication. Biochem Pharmacol 38:1709–1715
Moreira GE, Rosa GJM, Barros SBM et al (2001a) Antioxidant defense in rat brain regions after developmental lead exposure. Toxicology 169:145–151
Moreira GE, Vassilief I, Vassillief VS (2001b) Developmental lead exposure: behavioural alterations in the short and long term. Neurotoxicol Teratol 23:489–495
Nava-Ruíz C, Alcaraz-Zubeldia M, Méndez-Armenta M et al (2010) Nitric oxide synthase immunolocalization and expression in the rat hippocampus after sub-acute lead acetate exposure in rats. Exp Tox Pathol 62:311–316
Nehru B, Kanwar SS (2004) N-acetylcysteine exposure on lead-induced lipid peroxidative damage and oxidative defense system in brain regions of rats. Biol Trace Elem Res 101:257–264
O’Callaghan JP, Jensen KF (1992) Enhanced expression of glial fibrillary acidic protein and the cupric silver degeneration reaction can be used as sensitive and early indicators of neurotoxicity. Neurotoxicology 13:113–122
O’Dell TJ, Hawkins RD, Kandel ER et al (1991) Test of the roles of two diffusable substances in long-term potentiation: evidence for nitric oxide as a possible early retrograde messenger. Proc Natl Acad Sci 88:11285–11289
Oteiza PI, Verstraeten SV, Adonaylo VN (1995) Oxidative damage induced by metals without redox capacity in biological systems. Ci Cult 47:330–335
Papanikolaou CN, Hatzidaki GE, Belivanis S et al (2005) Lead toxicity update. A brief review. Med Sci Monit 11:329–336
Pautz A, Art J, Hahn S et al (2010) Regulation of the expression of inducible nitric oxide synthase. Nitric Oxide 23:75–93
Riccio A, Alvania RS, Lonze BE et al (2006) A nitric oxide signaling pathway controls CREB-mediated gene expression in neurons. Mol Cell 21:283–294
Rice DC (1993) Lead induced changes in learning: evidence for behavioral mechanisms from experimental animals studies. Neurotoxicology 14:167–178
Rice DC (1996) Behavioral effects of lead: commonalities between experimental and epidemiologic data. Environ Health Perspect 104:337–351
Robinson GB, Reed GD (1992) Effect of MK-801 on the induction and subsequent decay of long-term potentiation in the unanesthetized rabbit hippocampal dentate gyrus. Brain Res 569:78–85
Santizo K, Baughman VL, Pelligrino DA (2000) Relative contributions from neuronal and endotelial nitric oxide synthases to regional cerebral blood flow changes during forebrain ischemia in rats. NeuroReport 11:1549–1553
Sasaki M, Gonzalez-Zulueta M, Huang H et al (2000) Dynamic regulation of neuronal NO synthase transcription by calcium influx through a CREB family transcription factor-dependent mechanism. PNAS 97:8617–8622
Selvin-Testa A, Capani F, Loidl CF et al (1997) The nitric oxide synthase expression of rat cortical and hippocampal neurons changes after early lead exposure. Neurosci Lett 236:75–78
Sharifi AM, Baniasadi S, Jorjani M, Rahimi F, Bakhshayesh M (2002) Investigation of acute lead poisoning on apoptosis in rat hippocampus in vivo. Neurosci Lett 329:45–48
Simonian NA, Coyle JT (1996) Oxidative stress in neurodegenerative diseases. Annu Rev Pharmacol Toxicol 36:83–106
Skoczynska A, Smolik R, Jelen M (1993) Lipid abnormalities in rats given small doses of lead. Arch Toxicol 97:200–204
Soltaninejad K, Kebriaeezadeh A, Minaiee B et al (2003) Biochemical and ultrastructural evidences for toxicity of lead through free radicals in rat brain. Hum Exp Toxicol 22:417–423
Song XJ, Wang ZB, Gan Q et al (2006) cAMP and cGMP contribute to sensory neuron hiperexcitability and hiperalgesia in rats with dorsal root ganglia compression. J Neurophysiol 95:479–492
Squire LR, Knowlton BJ (1995) Memory, hippocampus and brain systems. In: Gazzaniga KS (ed) The cognitive neurosciences, xx edn. MIT Press, Cambridge, pp 825–837
Stoltenburg-Didinger G, Pünder I, Peters B et al (1996) Glial fibrillary acidic protein and RNA expression in adult rat hippocampus following low-level lead exposure during development. Histochem Cell Biol 105:431–442
Strużyñska L, Bubko I, Walski M et al (2001) Astroglial reaction during the early phase of acute lead toxicity in the adult rat brain. Toxicology 165:121–131
Sun L, Zhan ZY, Hu J et al (2005) Potential association of lead exposure during early development of mice with alteration of hippocampus nitric oxide levels and learning memory. Biomed Environ Sci 18:375–378
Talarek S, Fidecka S (2003) Role of nitric oxide in anticonvulsant effects of benzodiazepines in mice. Pol J Pharmacol 55:181–191
Talavera CE, Condes LM, Martínez LG (2003) Aspectos sobre las funciones del óxido nítrico como mensajero celular en el sistema nervioso central. Salud Ment 26:42–50
Thorns V, Hansen L, Masliah E (1998) nNOS expressing neurons in the entorhinal cortex and hippocampus are affected in patients with Alzheimer’s disease. Exp Neurol 150:14–20
Tiffany-Castiglioni E (1993) Cell culture models for lead toxicity in neuronal and glial cells. Neurotoxicology 14:513–536
Tiffany-Castiglioni E, Qian Y (2001) Astroglia as metal depots: molecular mechanisms for metal accumulation, storage and release. Neurotoxicology 22:577–592
Tiffany-Castiglioni E, Sanabria E, Wu E et al (1990) Lead uptake by cultured astroglia. In vitro Cell Develop Bio 26:66A
Toda N, Okamura T (2003) The pharmacology of nitric oxide in the peripheral nervous system of blood vessels. Pharmacol Rev 57:315–338
Toscano CD, Guilarte RT (2005) Lead neurotoxicity: from exposure to molecular effects. Brain Res Rev 49:529–554
Valko M, Rhodes CJ, Moncol J et al (2006) Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact 160:1–40
Van Den Berg KJ, Lammers JHCM, Hoogendijk EMG et al (1996) Changes in regional brain GFAP levels and behavioral functioning following subchronic lead acetate exposure in adult rats. Neurotoxicology 17:725–734
Villeda-Hernández J, Barroso-Moguel R, Méndez-Armenta M et al (2001) Enhanced brain regional lipid peroxidation in developing rats exposed to low level lead acetate. Brain Res Bull 55:247–251
Villeda-Hernández J, Méndez-Armenta M, Barroso-Moguel R et al (2006) Morphometric analysis of brain lesions in rat fetuses prenatally exposed to low-level lead acetate: correlation with lipid peroxidation. Histol Histopathol 21:609–617
Wang Y, Newton DC, Marsden PA (1999) Neuronal NOS: gene structure mRNA diversity, and functional relevance. Crit Rev Neurobiol 13:21–43
Wang J, Wu J, Zhang Z (2006) Oxidative stress in mouse brain exposed to lead. Ann Occup Hyg 50:405–409
Weiss SW, Albers DS, Iadarola MJ et al (1998) NMDA R1 glutamate receptor subunit isoforms in neostriatal, neocortical, and hippocampal nitric oxide synthase neurons. J Neurosci 18:1725–1734
White LD, Cory-Slechta DA, Gilbert ME et al (2007) New and evolving concepts in the neurotoxicology of lead. Toxicol Appl Pharmacol 225:1–27
Wiencken AE, Casagrande VA (1999) Endothelial nitric oxide synthase (eNOS) in astrocytes another source of nitric oxide in neocortex. Glia 26:280–290
Xu YZ, Ruan DY, Wu Y et al (1998) Nitric oxide affects LTP in area CA1 and CA3 of the hippocampus in low-level lead-exposed rat. Neurotoxicol Teratol 20:69–73
Yang Y, Ma Y, Ni L et al (2003) Lead exposure through gestation-only caused long-term learning/memory deficits in young adult offspring. Exp Neurol 184:489–495
Zengh W, Aschner M, Ghersi-Egea JF (2003) Brain barrier system: a new frontier in metal neurotoxicological research. Toxicol Appl Pharmacol 192:1–11
Zhang S, Chen J, Huang S (1998) Spatial learning and memory induce up-regulation of nitric oxide-producing neurons in rat brain. Brain Res 801:101–106
Zhao Y, Brandish PE, Ballou PD et al (1999) A molecular basis for nitric oxide sensing by soluble guaylate cyclise. Proc Natl Acad Sci 96:14753–14758
Zhou I, Zhu DY (2009) Neuronal nitric oxide synthase: structure, subcellular localization, regulation, and clinical implications. Nitric Oxide 20:223–230
Zhu ZW, Yang RL, Dong GJ et al (2005) Study on the neurotoxic effects of low-level lead exposure in rats. J Zhejiang Univ Sci B 6:686–692
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nava-Ruiz, C., Méndez-Armenta, M. & Ríos, C. Lead neurotoxicity: effects on brain nitric oxide synthase. J Mol Hist 43, 553–563 (2012). https://doi.org/10.1007/s10735-012-9414-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10735-012-9414-2