
Abstract
Dendrocalamus strictus, popularly known as “lathi baans”, is a multipurpose bamboo species known for its manifold domestic and industrial applications. In the current scenario, the extraction of raw material (culms) has surpassed the available growing stock, which puts the accessible genetic resource under pressure. Despite the commercial significance and overexploitation, population genetics of this valuable species is poorly studied, mainly due to the paucity of genomic information and marker resources. Moreover, polyploid genome structure obstructs the usage of robust codominant markers like simple sequence repeats (SSRs) for genetic analysis. Hence, the present study was conducted to develop genome sequence information as well as de novo SSRs in D. strictus. About 15 Gb sequence data was generated by sequencing 103.95 million reads using the Illumina protocol, which were further assembled into 902,453 contigs with 47.15% GC content and a 712 bp N50 value. A total of 39,473 microsatellite repeats were identified, wherein di-nucleotides were the most abundant (64.42%), followed by tri (29.66%), tetra (4.53%), penta (1.16%), and hexa-repeats (0.24%). Primer pairs were developed for 20,606 SSRs, and 250 of these were selected for validation. Through PCR amplification in 20 individuals, 69 SSRs displayed polymorphic banding pattern, of which 43 were single-locus and 26 were multi-locus SSRs. By comparison, both primer sets were reliable, giving similar results in analysed individuals. The available sequence data and unique single-locus SSR markers have the potential to improve our understanding towards D. strictus genomic background. The study also highlights the usability of identified SSRs in polyploids.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Bamboo is one of the strongest and fastest growing arborescent plants on earth. India is home to 125 native and 11 exotic bamboo species with an area cover of about 15 million hectares (FSI 2021). Among these, Dendrocalamus strictus (Roxb.) Nees occupies the maximum bamboo area (53%), and is the most commonly and commercially utilised bamboo (Das et al. 2017). Owing to its versatile utility, spanning from domestic products to rural housing and raw materials for cottage industries, it has been appreciated as one of the worthwhile alternatives to wood. Its raw material is extensively utilised in the pulp and paper industries of India, and about 200,000 tons of paper pulp per year is produced exclusively from bamboos (Tripathi et al. 2018). Additionally, its leaves are rich in natural antioxidants, and the young shoots are edible having high nutritional qualities (Yeasmin et al. 2015).
Although this plant has myriad uses and an adaptable nature to flourish in diverse edaphic and climatic conditions, its production is still low to match the growing demand. Unavailability of quality planting material, difficult propagation, unpredictable flowering (30–45 years), poor seed sets, and a short seedling life span are the key issues in commercial cultivation (Goyal et al. 2015). Notably, negligent extraction may cause a swift decline in stand structure as well as underlying genetic processes in artificially cultivated and naturally grown populations. Though no baseline knowledge of population genetics is available in this species, some research on other Indian bamboo species, namely Melocanna baccifera (Nilkanta et al. 2017) and D. hamiltonii (Meena et al. 2019), emphasised the urgent requirement of conserving the existing populations after reporting low genetic diversity. Besides, a comprehensive analysis of genetic diversity, gene flow, population structure, and phylogenetic research becomes crucial to develop effective conservation and management strategies for bamboo genetic resources. Emerging approaches like association mapping (AM), which exploits historical recombination of genes for trait-association analysis in the natural population, open up new prospects for marker-assisted selection (MAS) and genetic improvement of bamboo (Bhandawat et al. 2016). Through this approach, a marker linked to traits of commercial importance could be identified for further selection of superior genotypes. But the accomplishment of such targets requires a huge number of markers, which are limited in this species.
Microsatellites, also known as simple sequence repeats (SSRs), are one of the most valuable and insightful molecular marker techniques. Their abundance in the genome, co-dominance, high polymorphism, locus specificity, high reproducibility, and cross-transferability between close relatives make them the marker of choice for various genetic approaches (Taheri et al. 2018). The initial wave of marker-based research failed to utilize SSRs in bamboos due to lack of genomic data. Till then, most genetic research employed random primer based non-specific markers like Amplified Fragment Length Polymorphism (AFLP), Inter Simple Sequence Repeats (ISSR) and Random Amplified Polymorphic DNA (RAPD). Numerous SSRs of closely related species were also tested for cross-amplification; for instance, SSRs of rice, sugarcane, Bambusa arundinacea, and D. latiflorus were successfully amplified in D. strictus (Sharma et al. 2008; Bhandawat et al. 2014). But in the present scenario, research methodologies are focused on developing de novo SSRs due to the availability of advanced and cost-effective sequencing technology. Notably, existing SSR marker resources are inadequate to draw definite genotyping conclusions across the bamboo complex. Additionally, D. strictus has a hexaploid genome (2n = 6X = 72) (Thakur et al. 2015), and genotyping using SSRs could generate multiple alleles at some loci. With the given facts, such data are difficult to score and analyse. In most research, these SSRs are generally analysed as non-allelic data like dominant markers, where we lose several advantageous genetic characteristics expected with codominant SSR markers.
To address the above issues, present study was aimed to (1) generate genome sequence information for D. strictus through high throughput next generation sequencing (NGS) technology, (2) develop and characterize de novo microsatellite markers in D. strictus, and (3) identify single-locus SSRs (SL-SSRs). Our study also provides a comparative analysis of SL-SSRs with multi-locus SSRs (ML-SSRs), which could expand the understanding of SSR usage in polyploids.
Materials and methods
Sample collection and genomic DNA extraction
Twenty individuals were marked in the germplasm repository of D. strictus at Forest Research Institute, Dehradun, for sample collection, which comprised five individuals each from four geographical locations, viz., Tamil Nadu (DSTN), Telangana (DSTS), Haryana (DSHR), and Uttar Pradesh (DSUP) (Table 1). Young foliage samples were collected from selected individuals and surface sterilized prior to DNA extraction. Total genomic DNA was isolated using the Doyle and Doyle (1990) protocol with slight modifications. Further, genomic DNA was subjected to quantitative and qualitative analysis using a Bio-photometer (Eppendorf) and gel electrophoresis (0.8% agarose), respectively.
Library construction and sequencing
For sequencing, juvenile leaf sample was harvested from a genotype of D. strictus (DSBR-10.38) present in the germplasm repository of Forest Research Institute, Dehradun (Uttarakhand, India), which was originally collected from Gaya (Bihar, India). Genomic DNA of the above genotype was subjected to preparing small fragment libraries with a mean fragment size of 350 bp using the Illumina protocol. Approximately 1 µg of genomic DNA was fragmented using Covaris M220, which was further imported into NEB NEXT DNA II Library prep kit for genomic library construction. The resulting data was checked quantitatively and qualitatively using Qubit HS dsDNA assay and Agilent Bioanalyzer DNA HS assay, respectively. The QC passed two paired-end libraries were sequenced by Clevergene Biocorp Private Limited, Bengaluru, Karnataka, using HiSeq X Ten System (Illumina). Softwares FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and MultiQC (Ewels et al. 2016) were employed to analyse the quality of raw sequence data for base call quality distribution, % bases above Q20 and Q30, % GC, and adapter contaminations. The raw sequence reads were further processed to remove adapters and low-quality sequences via fastp (Chen et al. 2018). Clean reads were subsequently assembled into contigs with four k-mer sizes, 49, 67, 99, and 121, using Abyss ver 2.0.2 (Simpson et al. 2009). Besides, another assembly was also generated through Megahit ver 1.1.3 (Li et al. 2015). The final assembly for SSR mining was selected based on the recorded quality parameters, viz., overall alignment rate, number of contigs, proportion of reads mapped, N50, L50, etc.
SSR mining
The SSR loci were mined out from assembled contigs using Perl scripts-based program MIcroSAtellite (MISA) identification tool (Beier et al. 2017). Afterwards, the SSRs with optimal flanking sequences on both ends were scanned for primer designing using software Primer 3 (https://pubmed.ncbi.nlm.nih.gov/22730293/) with default parameters. A Linux-based Krait tool was used to estimate the relative abundance (loci Mb−1) and relative density (bp Mb−1) of SSRs (Du et al. 2018). Further, the potential utility of SSRs was unveiled by assigning putative functions to different SSR sequences, for which corresponding contigs were subjected to a sequence similarity search against the non-redundant (nr) protein database through NCBI BLASTx (Johnson et al. 2008; https://blast.ncbi.nlm.nih.gov/Blast.cgi).
Validation and characterization of SSRs
A panel of 250 SSRs covering all repeat classes was selected and tested for their polymerase chain reaction (PCR) amplification with the genomic DNA of the sampled individual DSBR-10.38 using a thermal cycler (Eppendorf Mastercycler Nexus). In the first PCR trial, the annealing temperature was optimized for each primer pair via gradient PCR, where a range of annealing temperatures (Expected Tm ± 5 °C) was used to get the best amplification product. The PCR reactions were performed in a total volume of 15 µL mixture containing 60 ng of template DNA, 1.5 µL of 10× PCR buffer, 1.75 mM MgCl2, 0.2 mM dNTPs, 0.1 µM of each forward and reverse primer, 0.6 units of Taq DNA polymerase, and nuclease-free sterile water. Each amplification reaction started with an initial denaturation at 95 °C for 5 min, followed by 35 cycles of denaturation at 94 °C for 1 min, annealing at primer-specific expected range of temperature for 1 min, extension at 72 °C for 1 min, and final extension at 72 °C for 10 min. The PCR products were resolved on a 2% agarose gel buffered with 1× TBE (Tris/Borate/EDTA). Primer pairs showing positive amplification were further characterised for polymorphism and other marker characteristics by subjecting them to PCR amplification in 20 genotypes of 4 distinct provenances (Table 1). Amplified products were separated on 4% high-resolution agarose gel (Sigma-Aldrich) and visualised under gel documentation system (UVP). The primer pairs showing allele size variation among the individuals were marked as polymorphic and recommended for further use.
Statistical analysis
The band profile generated by each polymorphic SSR was scored manually. Owed to the polyploid genome, several SSRs showed multi-locus amplification, resulting in multiple bands in a single individual. However, many SSR loci were perfectly amplifying at a single locus. The SSR loci showing a maximum of two bands per genotype were designated as single-locus SSRs (SL-SSRs), and the SSRs with multiple bands per genotype were designated as multi-locus SSRs (ML-SSRs). The SL-SSRs were scored as allelic data and analysed as codominant markers, while the ML-SSRs were scored as non-allelic data and analysed as dominant markers. Accordingly, both data sets were analysed separately to calculate various marker characteristics using different software programs. PowerMarker ver 3.25 (Liu and Muse 2005) was used to evaluate polymorphism information content (PIC). Subsequently, the marker data was used to determine various population genetic parameters such as numbers of alleles per locus (Na), numbers of effective alleles (Ne), observed heterozygosity (Ho), expected heterozygosity (He), measures of genetic differentiation (FST, GST and PhiPT), inbreeding coefficient (FIS), and number of private bands (bands unique to a particular population) using software GenAlEx ver 6.5 (Peakall and Smouse 2012). Analysis of molecular variance (AMOVA) between and within the provenances was also conducted via GenAlEx. Software POPGENE ver 1.31 (Yeh et al. 1999) was used to estimate Nei’s genetic diversity (h) and GST for ML-SSRs and gene flow (Nm) for both marker sets. Further cluster analysis was carried out using UPGMA (Unweighted Pair Group Method with Arithmetic Mean) and SAHN clustering tool in NTSYS-pc ver 2.10 (Rohlf 1998). The data was eventually used to derive the population genetic structure of 20 genotypes by software STRUCTURE ver 2.2 (Pritchard et al. 2000), where the simulations were run ten times for each set K values from 1 to 10 with 150,000 Markov Chain Monte Carlo (MCMC) sampling runs after a burn-in period of 150,000. The optimum value of K was determined using a web-based program STRUCTURE HARVESTER ver 0.6.92 (Earl and vonHoldt 2012; http://taylor0.biology.ucla.edu/structureHarvester/).
Results
A total of 103.95 million raw reads were generated through paired-end low-depth genome sequencing, with an average read length of 150 bp. The quality parameters, such as GC content, bases above Q20, and bases above Q30, were recorded as 47.15%, 99.92%, and 97.34%, respectively (Table 2). After filtration and trimming, the clean reads with a size of more than 70 bp were de novo assembled into contigs with different k-mer sizes, such as 49, 67, 99, and 121 bases. Assemblies with k-mer size 99 and 121 had very low genome size when compared to those with k-mer 49 and 67. Hence, reads were mapped back to these two large assemblies. However, most of the contigs in both assemblies were obtained below 1000 bp, and the proportion of reads mapped back to the genome was also low (Table 3). Therefore, the reads were again de novo assembled into another algorithm, i.e., Megahit. Comparatively, Megahit assembly was more robust, with good number and size of contigs, a better N50 (712 bp), % mapped reads (74%), and assembly length (~ 462 Mb). In total, 902,453 contigs were obtained, of which 864,474 contigs were attained with length between 200 and 1000 bp and 37,979 contigs more than 1000 bp. Owed to the low contig N50, the proportion of longer contigs (≥ 5000 bp) was very low in our genome assembly. After scanning the genome assembly for microsatellite repeats, a total of 39,473 SSRs were mined out. The raw genome sequence data generated herein was deposited into the Sequence Read Archive (SRA) database under accession PRJNA839537.
Frequency distribution of SSRs in genome assembly
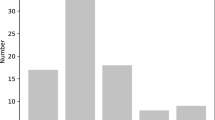
Among different repeat classes, di-nucleotides were the most predominantly occurring repeats in the genome, accounting for 64.42%, followed by tri- (29.66%), tetra- (4.53%), penta- (1.16%), and hexa-nucleotide repeats (0.24%) (Fig. 1a). In the di-repeats, AG/CT and AC/GT were the most dominant motifs compared with AT/AT and CG/CG, and CCG/GCC was the most abundant tri-repeat motif, followed by AGG/CCT and AGC/CTG. However, AT-rich motifs were more frequent in larger repeat classes, such as ACAT/ATGT in tetra- and AAAAG/CTTTT in penta-repeats (Fig. 1b–e). In order to obtain a comprehensive knowledge of the SSR distribution in the genome, they were characterized for their relative abundance (loci/Mb) and density (bp/Mb), which were maximum for di-nucleotides (35.57 loci Mb−1, 687.25 bp Mb−1), followed by tri-, mono-, tetra-, penta-, and hexa-repeats (Fig. 1f). Furthermore, the BLAST hits inferred the functional resemblance of many repeat motifs with different protein families. Digitaria exilis, Oryza sativa, Brachypodium distachyon, Triticum dicoccoides, Zea mays, Panicum hallii, Asparagus officinalis, and Panicum miliaceum were among the top hit species with minimum E values (Table 4).

Distribution and frequency of SSRs in genome of D. strictus: a Frequency of repeat types; b–e Frequency of repeat motifs; f Relative abundance and density of SSRs in genome
Primer designing and verification of the SSRs
Primer pairs were successfully designed for 20,606 SSR loci having flanking sequences ≥ 100 bp and repeat length ≥ 12 bp. The SSR loci were given the prefix “DSGS” to stand for “Dendrocalamus strictus Genomic SSR”. A comprehensive list of SSR loci along with their primer characteristics is given in Supplementary Table 1. Further, a panel of 250 SSR primer pairs was synthesized for their validation, in which 40% were selected for tri- and tetra-repeats, 24% for di-repeats, 16% for penta- and hexa-repeats, and the remaining 20% were for compound repeats. Of these, 215 SSRs were amplified successfully, yielding a clear band within the expected size range. Afterwards, polymorphism survey depicted 69 SSRs displaying different sized bands across the twenty test individuals. In congruence with the fact that D. strictus is a polyploid, we observed 26 polymorphic SSRs generating multiple alleles (≥ 2 bands per locus) in most test individuals, as shown in Fig. 2b, and these were considered ML-SSRs. However, the remaining 43 SSRs showed a standard diploid like banding pattern as shown in Fig. 2a (presenting 1 to 2 bands per locus), and therefore these were considered SL-SSRs. Both the data sets generated by SL-SSRs and ML-SSRs were analysed to reveal their genetic characteristics by treating them as allelic and non-allelic markers, respectively.

Representative image of PCR amplification of D. strictus genotypes using a DSGS-16532 (SL-SSR) and b DSGS-20351 (multi-locus simple sequence repeats); where, Lane M: 100 bp DNA ladder; Lane 1 to 20: individual genotypes of four provenances; and DSTN, DSTS, DSHR and DSUP represents individuals from Tamil Nadu, Telangana, Haryana and Uttar Pradesh, respectively
SL-SSRs genetic characterization
By genotyping 20 individuals, 43 SL-SSRs generated a total of 165 alleles, ranging from 2 to 7 alleles per locus. The highest seven alleles were displayed by DSGS-19663. The detailed marker characteristics are given in Table 5. All the evaluated SSRs were highly polymorphic, with a PIC value ranging from 0.048 (DSGS-16968) to 0.783 (DSGS-14515). The observed heterozygosity ranged between 0 and 1, with a mean value of 0.221. Likewise, expected heterozygosity ranged between 0.045 and 0.645, with an average of 0.312. Excess of homozygotes is evident at many SSR loci with a mean inbreeding coefficient value 0.306.
Further, AMOVA showed that 70% of the genetic variation was attributable to differences within individuals from the same geographical region, and 30% variation occurred among individuals from different regions (Supplementary Table 2a). Overall, SSR marker loci indicated high genetic differentiation (FST= 0.295) and a moderate level of gene flow (Nm = 0.486). Additionally, genetic relationship among the 20 test individuals was evaluated through UPGMA clustering. The UPGMA dendrogram (Fig. 3a) clustered the genotypes into two distinct major groups (Gp) at similarity coefficient 0.65, where Gp I had 15 genotypes belonging to DSTN, DSTS, and DSHR, while the individuals of DSUP formed a separate group (Gp II). Group 1 (Gp I) was further subdivided into 2 subgroups (SbGp), i.e., SbGp-Ia (5 genotypes representing DSTN) and SbGp-Ib (10 genotypes representing DSTS and DSHR). In agreement to the above observations, structure analysis revealed an optimum K value of 3, indicating three distinct groups (Supplementary Fig. 1a). The bar plot (Fig. 4a) shows the pattern and extent of genetic admixing between individuals of different regions, and the overall topology is found in congruence to the UPGMA dendrogram. Though the analyses provide a fair idea of genetic differentiation and structuring, results could not be used for conservation implications because the number of samples per geographic region was not adequate.

The UPGMA dendrogram unbiased measure of genetic distance among 20 genotypes of four provenances of D. strictus through single locus simple sequence repeats (a) and multi-locus simple sequence repeats (b). DSTN, DSTS, DSHR and DSUP represent individuals from Tamil Nadu, Telangana, Haryana and Uttar Pradesh, respectively

Bar plot for estimated population Q-matrix at a K = 3 through single locus simple sequence repeats and b K = 4 through multilocus simple sequence repeats for all 20 genotypes of D. strictus. Individual samples are represented by coloured bars and vertical lines separate different provenances
ML-SSRs genetic characterization
PCR amplification of 20 individuals with 26 ML-SSRs displayed 3–12 bands per marker locus. Considering each band as an individual locus, the data was scored in binary matrix and analysed as dominant marker. The PIC value varied from 0.114 (DSGS-18301) to 0.436 (DSGS-19467), with an average value of 0.305. The average values of Shannon information index (I), Nei’s genetic diversity (h), Gene flow (Nm), GST, and PhiPT were 0.232, 0.262, 0.715, 0.412, and 0.366, respectively (Supplementary Table 3).
Similar to SL-SSRs, AMOVA displayed maximum 63% variation within individuals of the same provenance and remaining 37% variation was detected among individuals of different provenances (Supplementary Table 2b). The UPGMA dengrogram based on Jaccard’s similarity coefficient (Fig. 3b) showed two major groups: Gp I consisted of 10 genotypes from DSTN and DSTS, and Gp II had genotypes from DSUP and DSHR. Further sub-clustering was uneven, where SbGpIa was represented by 7 genotypes from DSTN and DSTS, SbGpIb had 3 genotypes from DSTS, SbGpIIa showed 6 genotypes representing DSTS and DSHR, and SbGpIIb was solely determined to 4 genotypes from DSUP. Also, the STRUCTURE analysis revealed four major clusters (Supplementary Fig. 1b), and the genetic admixing among individuals at K = 4 is displayed as bar plot (Fig. 4b).
Discussion
In the current scenario, a complete genome sequence is available in many crops belonging to the family Poaceae (Ramakrishnan et al. 2020); however, genome-level investigations in bamboo still remain limited. The draft genome of Moso bamboo by Peng et al. (2013) was the primary genome presented in the subfamily Bambusoideae, which paved the way forward for genomic research in bamboo. Sequencing of plant genome is challenging due to the complexity aroused by high rates of heterozygosity and repeats (Gore et al. 2009; Schatz et al. 2012), large gene families accompanied by compound gene content, and plentiful pseudogenes (Schnable et al. 2009). In addition, approximately 80% of plant species are polyploids (Meyers and Levin 2006). Nevertheless, the availability of cost-effective and rapid NGS technology has up-scaled the generation of massive high-quality sequence data and provide great opportunities to expand our understanding to complex genomes of several plants, including bamboo. Among various NGS platforms, the Illumina genome analyzer is one of the most widely utilized approaches used in plant genome sequencing and microsatellite identification (Taheri et al. 2018). Recently, this technology has been successfully utilized to develop microsatellite markers in a variety of species, viz., Populus pruinosa (Yang et al. 2017), Grevillea juniperina (Damerval et al. 2019), Rhodoleia championii (Huang Y et al. 2019; Huang C et al. 2019), Exbucklandia tonkinensis (Huang Y et al. 2019; Huang C et al. 2019), Salvadora oleoides (Bhandari et al. 2020), G. robusta (Dabral et al. 2021), and Platostoma palustre (Zheng et al. 2022). Similarly, genic and genomic SSRs have also been developed in some bamboo taxa, viz., Phyllostachys edulis (Zhao et al. 2015), P. violascens (Cai et al. 2019), and Drepanostachyum falcatum (Meena et al. 2021). However, limited genomic and marker information is available for the genus Dendrocalamus, which is widely distributed in the Indian subcontinent and possesses high commercial significance. Prior to our work, expressed sequence tags (ESTs)-based genic SSRs were developed in D. latiflorus (Bhandawat et al. 2016) and D. hamiltonii (Bhandawat et al. 2019). Later, these SSRs were also tested for their cross-transferability in 36 bamboo species, including D. strictus (Bhandawat et al. 2014; Tripathi 2019). Recently, de novo SSRs have been developed using a similar approach in D. longispathus (Meena et al. 2023).
Genome assembly and microsatellite distribution
Approximately 15 Gb raw sequence data was assembled into 902,453 contigs representing ~ 462 Mb genome with a GC content of 47.15% and a contig N50 value of 712 bp. Markedly, a large proportion of contigs (~ 96%) were obtained with a size less than 1000 bp. In general, the contig N50 value states that the contig length is equivalent to the size of the blocks needed to cover half of the assembled genome. Recent research conducted on G. robusta and D. longispathus revealed higher contig N50 (Dabral et al. 2021; Meena et al. 2023). Accordingly, a lower N50 value in our data indicates a fragmented genome assembly, which could be attributed to lesser genome coverage and polyploidy (Otto et al. 2008; Meena et al. 2021). For instance, the assembly (~ 462 Mb) obtained in this study covers only 17% of the estimated genome size of 2.7 Gb of its close relative, P. heterocycla (Peng et al. 2013). Genome assembly was further complicated due to the higher ploidy level of D. strictus, which possesses a hexaploid genome (2n = 6X = 72). Hence, the assembly was not suitable for high-confidence annotation but good enough to unravel the basic knowledge of the genomic structure and functions in D. strictus, like SSR distribution, which was the ultimate goal of this study. Due to inadequate genomic coverage, it may not be feasible to detect all the genomic microsatellite repeats, but it gives a reliable estimate of their frequency in the genome (Otto et al. 2008).
In total, 39,473 microsatellite repeats were identified in our genome assembly. From all identified SSRs, the di-nucleotides were the most abundant repeats, followed by tri-, mono-, tetra-, penta-, and hexa-nucleotide repeats, respectively. The microsatellite frequency and distribution were found to be in congruence with different plant taxa, including bamboo, namely D. longispathus and D. falcatum (Meena et al. 2021, 2023; Zheng et al. 2022). However, our results differed from some other bamboo species, namely D. latiflorus (Bhandawat et al. 2016), D. hamiltonii (Bhandawat et al. 2019) and P. violascens (Cai et al. 2019), where the genome sequence was dominated by tri-nucleotides. The difference observed in frequency of di- and tri-repeats could be attributed to the approaches used for SSR development, i.e., random genome sequencing or transcriptome sequencing. For instance, mono- and di-repeats are the most common in the genome, but the coding region is dominated by tri-repeats (Bhandawat et al. 2016; Cai et al. 2019).
Beside the five bamboo species mentioned above, SSR characteristics were further compared with Moso bamboo (P. edulis) and some other members of the family Poaceae, viz., T. aestivum, Z. mays, B. distachyon, and Sorghum biocolor (Zhao et al. 2015). AG/CT (65.09%) was the most widespread di-repeat, which was consistent with previous research in D. latiflorus (75.03%), P. edulis (41.16%), Z. mays (47.74%), and B. distachyon (50.72%), whereas AT/AT was more common in S. biocolor (54.40%). For the tri-repeat motifs, CCG/CGG (28.15%) was the most predominant among all compared species except P. edulis, where AAG/CTT was plentiful. Interestingly, although CCG/CGG is a ubiquitous tri-repeat among monocotyledons, but its occurrence is rare in dicotyledons (Varshney et al. 2002; You et al. 2015), which may be due to high GC content and codon biasness in monocots (Morgante et al. 2002; Zhao et al. 2015). A wide diversity in repeat motifs was seen in tetra-, penta-, and hexa-nucleotide repeats; however, their overall genome contribution was less than 10%.
Consequently, the proportion of microsatellites decreases with an increase in repeat motif length. In general, the frequency of SSR motif types was observed to be conserved among the Poaceae species. Differences in frequencies of particular SSR repeat motifs may perhaps have an evolutionary function in plants, which needs to be further investigated.
Selection of SSRs, validation and characterization for polymorphism
Generally, the frequency of larger repeat motifs is lower in the genome, but the markers based on these are more reliable because they are less prone to amplification errors like SSR stuttering (Liljegren et al. 2016). Additionally, sequences with a larger repeat length are more polymorphic due to the higher mutation rate imposed by replication slippage (Chistiakov et al. 2006; Kostro-Ambroziak et al. 2020). Thus, SSR primers were carefully chosen for validation based on the number of repeats and GC content. As a result, 86% of selected SSRs were successfully amplified, yielding products within the expected size range. It also indicates the reliability of genomic assembly and SSRs. However, few SSRs failed to amplify, which could be explained by the presence of introns within the locus, splice sites across primers, or point mutations in primer binding sites.
Furthermore, the efficacy of microsatellite markers in polyploid D. strictus was investigated by scoring them as codominant and dominant data sets in accordance to an earlier study by Meena et al. (2020) in D. hamiltonii. Generally, SL-SSRs are considered more informative and robust than ML-SSRs. As indicated in this study, SL-SSRs give information about heterozygosity, which is not possible with ML-SSRs. Comparatively, the mean PIC value (0.486) calculated for SL-SSR in our study was equivalent to a recent study conducted in D. longispathus (PIC = 0.50) using SSR markers (Meena et al. 2023). Notably, DSGS-14515, DSGS-16022, DSGS-16583, DSGS-17703, DSGS-19198, and DSGS-19663 were distinguished as most informative with PIC ≥ 0.70, and approximately 51% of SL-SSRs were identified with PIC ≥ 0.50, reflecting the hyper-variable nature of tested SL-SSRs. Nonetheless, ML-SSRs produced a huge number of reproducible bands (3–12 bands per genotype), which makes them more desirable than the other less reproducible dominant markers like RAPD. The overall PIC analysed in both datasets was higher when compared to RAPD markers utilised by Das et al. (2017) in D. strictus (PIC range = 0.27–0.31). Therefore, usage of both types of SSRs in polyploids is highly advantageous.
Genetic diversity analysis
Understanding genetic diversity within and among populations has always been crucial for tree conservation and improvement programs. Here, molecular markers play an essential role in determining heterozygosity, gene flow, genetic differentiation, and structuring. The quantitative results of the allelic and non-allelic data sets are incomparable due to different measures of importance. For instance, observed and expected heterozygosity (Ho and He) are considered key indicators of genetic diversity in allelic data analysis, while Shannon information index (I) and Nei’s genetic diversity (h) are considered important measures of dominant data analysis (Meena et al. 2020). However, their outputs can be first interpreted and then compared based on non-quantitative measures. Before our work, a genetic study based on isozymes conducted over 640 individuals of D. strictus from 16 sites in the Central Western Ghats revealed an overall low genetic diversity (h = 0.197) (Ravikanth et al. 2008). In contrast, a recent study investigated 369 individuals from the germplasm repository of D. strictus, FRI, Dehradun and reported higher genetic diversity (He = 0.769) using cross-transferable SSRs (Tripathi 2019). The species-specific SL-SSRs (Ho = 0.221, He = 0.312) and ML-SSRs (I = 0.232, h = 0.262) tested in the current study infer a moderate level of genetic diversity, which was similar to the other research in different species of genus Dendrocalamus, namely D. longispathus (Ho = 0.277, He = 0.480) using species-specific SSR markers (Meena et al. 2023), D. strictus (h = 0.261, I = 0.403) using RAPD markers (Das et al. 2017), D. giganteus (He = 0.2978), and D. membranaceus (h = 0.219, I = 0.349) using ISSR markers (Tian et al. 2012; Yang et al. 2012). The majority (86%) of our SL-SSR markers showed overall He ≥ 0.10, and 16% displayed He ≥ 0.70. Previous research described that a marker is polymorphic if He ≥ 0.10 and highly polymorphic if He ≥ 0.70 (Ott 1992; Kullan et al. 2016).
Additionally, high genetic differentiation (FST = 0.295) and moderate gene flow (Nm = 0.486) were observed using SL-SSRs. Nevertheless, similar results were elucidated using different measures for ML-SSRs (PhiPT = 0.366, Nm = 0.715). According to Wright (1978), FST > 0.25 and Nm lying between 0.99 and 0.250 represent high differentiation and modest gene flow, respectively (Cheng et al. 2020). Another FST analogue, i.e., GST was also estimated to facilitate comparison of our findings (GST = 0.412) with other research. Interestingly, previous research by Ravikanth et al. (2008) using isozymes (GST = 0.27), and Das et al. (2017) using RAPD markers (GST = 0.49, Nm = 0.51) also reported high genetic differentiation and moderate gene flow in D. strictus. Similarly, moderate to high genetic differentiation has also been reported in other bamboo species, namely Ochlandra travancorica (Nag et al. 2013), M. baccifera (Nilkanta et al. 2017), Kuruna debilis (Attigala et al. 2017), D. sinicus (Yang et al. 2018), D. hamiltonii (Meena et al. 2019), and D. longispathus (Meena et al. 2023). This may be attributed to cross pollinating behaviour, protogynous mechanism and gregarious flowering coupled with a high degree of floral asynchrony across the provenances. Positive value of inbreeding coefficient (FIS= 0.306) suggests deficit of heterozygotes in the sampled individuals.
The above outcomes were also supported by AMOVA, where maximum genetic variation remained confined among the individuals of the same geographical region. Bamboos generally have a prolonged vegetative phase of 20–150 years (Ma et al. 2013), and many investigations suggested that genetic variation is least among populations in such long-lived and out-crossing species (Nybom 2004; Meena et al. 2019; Oumer et al. 2020). The UPGMA dendrogram and structure analysis further validated the results. The delta K value (K = 3 and 4) fosters to justify the meagre genetic exchange and slight admixing between different gene pools. The contrasting result of structure could be ascribed to analysis performed on lesser individuals per provenance, which can be further investigated by increasing the sample number. However, with K = 3, it was observed that DSTS and DSHR were significantly forming a cluster together. Haryana has the least bamboo-bearing area (39 sq km) in India (FSI 2021). Moreover, the majority of plantations in India are being carried out using seeds obtained from unknown sources with no particular mechanisms to certify the productivity credentials of their source (FRI 2014; Tewari et al. 2019). So, there might be a possibility that DSHR had been planted from source material obtained from Telangana (DSTS) gene pool. However, due to the lack of an adequate number of samples per population or per provenance, no significant structuring or clustering can be confirmed.
Conclusions
NGS offers quality data combined with affordable costs, improved data handling capabilities, increased computational power, and efficient bioinformatics analysis tools. The study presents first de novo genome sequence information generated in D. strictus. The large set of developed markers, particularly the subset of SL-SSR that has been validated, is highly valuable. These primer pairs could help in planning conservation and open up new opportunities for genetic analysis, AM and MAS in D. strictus, along with other related species. Furthermore, the present study promotes the utilisation of SL-SSRs in polyploids. However, based on the comparative analysis, it can be stated that in absence of SL-SSRs, ML-SSR markers can also be utilised in polyploid species. Though they may lose the property of co-dominance and fail to recognise heterozygosity and allelic richness, they still provide a greater advantage in reproducibility over other prevalent dominant markers.
Data availability
The additional data required to understand the manuscript is available in the supporting information of this manuscript and the raw sequence data are available in Sequence Read Archive (SRA) database of NCBI with accession number PRJNA839537.
References
Attigala L, Gallaher T, Nason J, Clark LG (2017) Genetic diversity and population structure of the threatened temperate woody bamboo Kuruna debilis (Poaceae: Bambusoideae: Arundinarieae) from Sri Lanka based on microsatellite analysis. J Natl Sci Found Sri 45(1):53–65
Beier S, Thiel T, Munch T, Scholz U, Mascher M (2017) MISA-web: a web server for microsatellite prediction. Bioinformatics 33:2583–2585. https://doi.org/10.1093/bioinformatics/btx198
Bhandari MS, Meena RK, Shamoon A, Saroj S, Kant R, Pandey S (2020) First de novo genome specific development, characterization and validation of simple sequence repeat (SSR) markers in genus Salvadora. Mol Biol Rep 47(9):6997–7008. https://doi.org/10.1007/s11033-020-05758-z
Bhandawat A, Sharma V, Sharma H, Sood A, Sharma RK (2014) Development and cross-transferability of functionally relevant micro-satellite markers in Dendrocalamus latiflorus and related bamboo species. J Genet 93:48–55. https://doi.org/10.1007/s12041-014-0377-9
Bhandawat A, Sharma V, Singh P, Seth R, Nag A, Kaur J, Sharma RK (2019) Discovery and utilization of EST-SSR marker resource for genetic diversity and population structure analyses of a subtropical bamboo, Dendrocalamus hamiltonii. Biochem Genet 57(5):652–672. https://doi.org/10.1007/s10528-019-09914-4
BhandawatA, Singh G, Raina AS, Kaur J, Sharma RK (2016) Development of genic SSR marker resource from RNA-Seq data in Dendrocalamus latiflorus. J Plant Biochem Biotechnol 25:179–190. https://doi.org/10.1007/s13562-015-0323-9
Cai K, Zhu L, Zhang K, Li L, Zhao Z, Zeng W, Lin X (2019) Development and characterization of EST-SSR markers from RNA-seq data in Phyllostachys violascens. Front Plant Sci 10(50):01–09. https://doi.org/10.3389/fpls.2019.00050
Chen S, Zhou Y, Chen Y, Gu J (2018) Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34:884–890. https://doi.org/10.1093/bioinformatics/bty560
Cheng J, Kao H, Dong S (2020) Population genetic structure and gene flow of rare and endangered Tetraena mongolica Maxim. Revealed by reduced representation sequencing. BMC Plant Biol 20(1):1–3. https://doi.org/10.1186/s12870-020-02594-y
Chistiakov DA, Hellemans B, Volckaert FA (2006) Microsatellites and their genomic distribution, evolution, function and applications: a review with special reference to fish genetics. Aquaculture 255(1–4):1–29
Dabral A, Shamoon A, Meena RK, Kant R, Pandey S, Ginwal HS, Bhandari MS (2021) Genome skimming-based simple sequence repeat (SSR) marker discovery and characterization in Grevillea robusta. Physiol Mol Biol Plants 27:1623–1638. https://doi.org/10.1007/s12298-021-01035-w
Damerval C, Citerne H, Conde e Silva N, Deveaux Y, Delannoy E, Joets J, Simonnet F, Staedler Y, Schönenberger J, Yansouni J, Le Guilloux M (2019) Unravelling the developmental and genetic mechanisms underpinning floral architecture in Proteaceae. Front Plant Sci 10:18. https://doi.org/10.3389/fpls.2019.00018
Das S, Singh YP, Negi YK, Shrivastav PC (2017) Genetic variability in different growth forms of Dendrocalamus strictus: Deogun revisited. N Z J Sci 47(1):1–2. https://doi.org/10.1186/s40490-017-0104-4
Doyle JJ, Doyle JL (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Du L, Zhang C, Liu Q, Zhang X, Yue B (2018) Krait: an ultrafast tool for genome-wide survey of microsatellites and primer design. Bioinformatics 34(4):681–683. https://doi.org/10.1093/bioinformatics/btx665
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Ewels P, Magnusson M, Lundin S, Kaller M (2016) MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 6(32):3047–3048. https://doi.org/10.1093/bioinformatics/btw35
FRI (2014) Bamboo productivity in forest and non-forest areas. In: Proceeding of national seminar, bamboo technical support group, Indian council of forestry research and education. Forest Research Institute, Dehradun
FSI (2021) India state of forest report. Forest Survey of India, Dehradun
Gore MA, Chia JM, Elshire RJ, Sun Q, Ersoz ES, Hurwitz BL, Peiffer JA, McMullen MD, Grills GS, Ross-Ibarra J, Ware DH, Buckler ES (2009) A first-generation haplotype map of maize. Science 326:1115–1117
Goyal AK, Pradhan S, Basistha BC, Sen A (2015) Micropropagation and assessment of genetic fidelity of Dendrocalamus strictus (Roxb.) Nees using RAPD and ISSR markers. 3Biotech 5:473–482. https://doi.org/10.1007/s13205-014-0244-7
Huang Y, Yin Q, Do VT, Meng K, Chen S, Liao B, Fan Q (2019) Development and characterization of genomic microsatellite markers in the tree species, Rhodoleia championii, R. parvipetala, and R. forrestii (Hamamelidaceae). Mol Biol Rep 46(6):6547–6556
Huang C, Yin Q, Khadka D, Meng K, Fan Q, Chen S, Liao W (2019) Identification and development of microsatellite (SSRs) makers of Exbucklandia (Hamamelidaceae) by high-throughput sequencing. Mol Biol Rep 46(3):3381–3386. https://doi.org/10.1007/s11033-019-04800-z
Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, Madden TL (2008) NCBI BLAST: a better web interface. Nucleic Acids Res 36(2):5–9. https://doi.org/10.1093/nar/gkn201
Kostro-Ambroziak A, Siekiera A, Czajkowska M, Pomorski JJ, Panagiotopoulou H (2020) Development of microsatellite loci and optimization of a multiplex assay for Latibulus argiolus (Hymenoptera: Ichneumonidae), the specialized parasitoid of paper wasps. Sci Rep 10(1):16068
Kullan AR, Kulkarni AV, Kumar RS, Rajkumar R (2016) Development of microsatellite markers and their use in genetic diversity and population structure analysis in Casuarina. Tree Genet Genomes 12:1–2
Li D, Liu C-M, Luo R, Sadakane K, Lam T-W (2015) MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de bruijn graph. Bioinformatics 31:1674–1676
Liljegren MM, de Muinck EJ, Trosvik P (2016) Microsatellite length scoring by single molecule real time sequencing—effects of sequence structure and PCR Regime. PLoS ONE 11(7):e0159232. https://doi.org/10.1371/journal.pone.0159232
Liu K, Muse SV (2005) PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21:2128–2129. https://doi.org/10.1093/bioinformatics/bti282
Ma QQ, Song HX, Zhou SQ, Yang WQ, Li DS, Chen JS (2013) Genetic structure in dwarf bamboo (Bashania fangiana) clonal populations with different genet ages. PLoS ONE 8(11): e78784. https://doi.org/10.1371/journal.pone.0078784
Meena RK, Bhandari MS, Ginwal HS (2020) Usage of microsatellite markers for characterization of polyploids: a case study in reference to hexaploid bamboo species. SilvaeGenetica 69(1):94–97. https://doi.org/10.2478/sg-2020-0013
Meena RK, Bhandhari MS, Barthwal S, Ginwal HS (2019) Genetic diversity and structure of Dendrocalamus hamiltonii natural metapopulation: a commercially important bamboo species of northeast Himalayas. 3 Biotech 9:60. https://doi.org/10.1007/s13205-019-1591-1
Meena RK, Kashyap P, Shamoon A, Dhyani P, Sharma H, Bhandari MS, Barthwal S, Ginwal HS (2023) Genome survey sequencing-based SSR marker development and their validation in Dendrocalamus longispathus. Funct Integr Genom 23(2):103. https://doi.org/10.1007/s10142-023-01033-z
Meena RK, Negi N, Uniyal N, Bhandari MS, Sharma R, Ginwal HS (2021) Genome skimming based STMS marker discovery and its validation in temperate hill bamboo, Drepanostachyum falcatum. J Genet 100(2):1–10. https://doi.org/10.1007/s12041-021-01273-7
Meyers LA, Levin DA (2006) On the abundance of polyploids in flowering plants. Evolution 60(6):1198–1206. https://doi.org/10.1111/j.0014-3820.2006.tb01198.x
Morgante M, Hanafey M, Powell W (2002) Microsatellites are preferentially associated with non repetitive DNA in plant genomes. Nat Genet 30:194–200
Nag A, Gupta P, Sharma V, Sood A, Ahuja PS, Sharma RK (2013) AFLP and RAPD based genetic diversity assessment of industrially important reed bamboo (Ochlandra travancorica Benth). J Plant Biochem Biotechnol 22:144–149. https://doi.org/10.1007/s13562-012-0114-5
Nilkanta H, Amom T, Tikendra L, Rahaman H, Nongdam P (2017) ISSR marker based population genetic study of Melocanna baccifera (Roxb.) Kurz: a commercially important bamboo of Manipur, North-East India. Scientifica. https://doi.org/10.1155/2017/37572
Nybom H (2004) Comparison of different nuclear DNA markers for estimating intraspecific genetic diversity in plants. Mol Ecol 13(5):1143–1155
Ott J (1992) Strategies for characterizing highly polymorphic markers in human gene mapping. Am J Hum Genet 51(2):283–290
Otto TD, Gomes LH, Alves-Ferreira M, de Miranda AB, Degrave WM (2008) ReRep: computational detection of repetitive sequences in genome survey sequences (GSS). BMC Bioinform 9:366. https://doi.org/10.1186/1471-2105-9-366
Oumer AO, Kifle D, Tileye F, Kassahun T, Durai J, Hyder MZ (2020) Genetic diversity, population structure, and gene flow analysis of lowland bamboo [Oxytenanthera abyssinica (A. Rich.) Munro] in Ethiopia. Ecol Evol 10(20):11217–11236. https://doi.org/10.1002/ece3.6762
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 28:2537–2539
Peng Z, Lu Y, Li L et al (2013) The draft genome of the fast-growing non-timber forest species moso bamboo (Phyllostachys heterocycla). Nat Genet 45(4):456–461. https://doi.org/10.1038/ng.2569
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155(2):945–959. https://doi.org/10.1093/genetics/155.2.945
Ramakrishnan M, Yrjälä K, Vinod KK, Sharma A, Cho J, Satheesh V, Zhou M (2020) Genetics and genomics of moso bamboo (Phyllostachys edulis): current status, future challenges, and biotechnological opportunities toward a sustainable bamboo industry. Food Energy Secur 9(4):e229. https://doi.org/10.1002/fes3.229
Ravikanth G, Nageswara Rao M, Singh D, Cheluvaraju J, Ganeshaiah KN, Uma Shaanker R (2008) Contrasting spatial patterns of distribution of genetic diversity in two important bamboo species in the Central Western Ghats, India. J Bamboo Rattan 7(12):41–52
Rohlf FJ (1998) NTSYS-pc: numerical taxonomy and multivariate analysis system, version 2.02e. Applied Biostatistics Inc., Exeter Software, Setauket
Schatz MC, Witkowski J, McCombie WR (2012) Current challenges in de novo plant genome sequencing and assembly. Genome biol 13(4):1–7
Schnable PS, Ware D, Fulton RS et al (2009) The B73 maize genome: complexity, diversity, and dynamics. Science 326:1112–1115
Sharma RK, Gupta P, Sharma V, Sood A, Mohapatra T, Ahuja PS (2008) Evaluation of rice and sugarcane SSR markers for phylogenetic and genetic diversity analyses in bamboo. Genome 51:91–103
Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJM, Birol İ (2009) ABySS: a parallel assembler for short read sequence data. Genome Res 19:1117–1123
Taheri S, Lee Abdullah T, Yusop MR, Hanafi MM, Sahebi M, Azizi P, Shamshiri RR (2018) Mining and development of novel SSR markers using next generation sequencing (NGS) data in plants. Molecules 23(2):399
Tewari S, Negi H, Kaushal R (2019) Status of bamboo in India. Int J Econ Plants 6(1):30–39
Thakur A, Barthwal S, Ginwal HS (2015) Genetic diversity in bamboos: conservation and improvement for productivity. In: Kaushik S, Singh YP, kumar D, Thapaliyal M, Barthwal S (eds) Bamboos in India. ENVIS Centre on Forestry, Dehradun, pp 131–146
Tian B, Yang HQ, Wong KM, Liu AZ, Ruan ZY (2012) ISSR analysis shows low genetic diversity versus high genetic differentiation for giant bamboo, Dendrocalamus giganteus (Poaceae: Bambusoideae), in China populations. Genet Resour Crop Evol 59:901–908. https://doi.org/10.1007/s10722-011-9732-3
Tripathi A (2019) Morpho-genetics analysis of provenance/source variation in Dendrocalmus strictus (Roxb.) Nees. Ph.D. Thesis, Forest Research Institute Deemed to be University, Dehradun
Tripathi SK, Mishra OP, Bhardwaj NK, Varadhan R (2018) Pulp and papermaking properties of bamboo species Melocanna baccifera. Cell Chem Technol 52(1–2):81–88
Varshney RK, Thiel T, Stein N, Langridge P, Graner A (2002) In silico analysis on frequency and distribution of microsatellites in ESTs of some cereal species. Cell Mol Biol Lett 7(2A):537–546
Wright S (1978) Evolution and the genetics of populations: a treatise in four volumes. Variability within and among natural populations, vol 4. University of Chicago Press, Chicago
Yang HQ, An MY, Gu ZJ, Tian B (2012) Genetic diversity and differentiation of Dendrocalamus membranaceus (Poaceae: Bambusoideae), a declining bamboo species in Yunnan, China, as based on inter-simple sequence repeat (ISSR) analysis. Int J Mol Sci 13(4):4446–4457
Yang JB, Dong YR, Wong KM, Gu ZJ, Yang HQ, Li DZ (2018) Genetic structure and differentiation in Dendrocalamus sinicus (Poaceae: Bambusoideae) populations provide insight into evolutionary history and speciation of woody bamboos. Sci Rep 8(1):16933. https://doi.org/10.1038/s41598-018-35269-8
Yang W, Wang K, Zhang J, Ma J, Liu J, Ma T (2017) The draft genome sequence of a desert tree Populus pruinosa. Gigascience 6(9):gix075. https://doi.org/10.1093/gigascience/gix075
Yeasmin L, Ali MN, Gantait S, Chakraborty S (2015) Bamboo: an overview on its genetic diversity and characterization. 3 Biotech 5(1):1–11. https://doi.org/10.1007/s13205-014-0201-5
Yeh FC, Yang RC, Boyles TBJ, Ye ZH, Mao JX (1999) POPGENE version 1.32: microsoft window-based freeware for population genetics analysis. Molecular Biology and Biotechnology Centre, University of Alberta, Edmonton
You Y, Liu D, Liu H, Zheng X, Diao Y, Huang X, Hu Z (2015) Development and characterisation of EST-SSR markers by transcriptome sequencing in taro (Colocasia esculenta (L.) Schoot). Mol Breed 35(6):1–1. https://doi.org/10.1007/s11032-015-0307-4
Zhao H, Yang L, Peng Z, Sun H, Yue X, Lou Y, Dong L, Wang L, Gao Z (2015) Developing genome-wide microsatellite markers of bamboo and their applications on molecular marker assisted taxonomy for accessions in the genus Phyllostachys. Sci Rep 5(1):1–10
Zheng Z, Zhang N, Huang Z et al (2022) Genome survey sequencing and characterization of simple sequence repeat (SSR) markers in Platostoma palustre (Blume) A.J.Paton (Chinese mesona). Sci Rep 12:355. https://doi.org/10.1038/s41598-021-04264-x
Acknowledgements
The authors are thankful to National Authority CAMPA, Ministry of Environment, Forest and Climate Change, New Delhi for their financial support and highly obliged to the Director, Forest Research Institute, Dehradun for providing research and laboratory facilities.
Funding
The work was financially supported by National Authority CAMPA, Ministry of Environment, Forest and Climate Change, New Delhi under National Compensatory Afforestation Fund (NCAF) scheme on ‘Strengthening Forestry Research for Ecological Sustainability and Productivity Enhancement’ vide letter number 72(XVIII)/2018/ICFRE(R)/RP/ProjectProposals/154.
Author information
Authors and Affiliations
Contributions
Project conceptualization, execution and manuscript writing were performed by SR, HSG, VR and SB; Original manuscript draft preparation, laboratory work, data analysis and inference were performed by SR and RKM; all the authors critically revised the final draft.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial conflict of interests to disclose.
Ethics approval
Not applicable.
Consent to participate
No minor participant was involved in the research.
Consent for publication
All the authors have approved the manuscript for submission and publication.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Rohilla, S., Ginwal, H.S., Rana, V. et al. Genome-wide discovery of single- and multi-locus simple sequence repeat markers and their characterization in Dendrocalamus strictus: a commercial polyploid bamboo species of India. Genet Resour Crop Evol 71, 2311–2339 (2024). https://doi.org/10.1007/s10722-023-01777-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10722-023-01777-4