
Abstract
Heat stress at later growth stage is one of the major limitations in achieving potential yield in barley. Incorporation of heat tolerance in the variety development process is an essential task that breeders would like to achieve by exploring new sources of genetic variability and their utilization. A better understanding of genetic variation in existing genotypes under heat stress is required to produce high yielding varieties with improved heat tolerance. An association mapping panel 2017 (AM2017), comprising of 316 genotypes, was evaluated under timely and late sown (heat stress) conditions for two consecutive crop seasons at Hisar in India during 2017–2019. Eight agro-morphological traits, mainly contributing to yield, were considered agro-morphological diversity study. Genetic diversity and population structure were explored by using the 50 K iSelect Illumina Barley SNP array. A set of 36,793 SNP markers, covering a genetic distance of 991.82 cM with an average marker density of 37.09 SNPs/cM, was obtained after quality filtration. The gene diversity (GD) and Polymorphic Information Content (PIC) at genome level was 0.362 and 0.289, respectively. In AM2017, two subpopulations were observed mainly due to the row types. Principal component analysis of agro-morphological traits revealed that days to heading and maturity along with spike length, spikelet number per spike and grain yield per plot were the most important traits in timely and late sown conditions. The information on agro-morphological genetic diversity under heat stress conditions will be useful in identifying heat tolerant genotypes for use in barley breeding programs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Barley (Hordeum vulgare L.), was one of the first domesticated food grain and grown in the Fertile Crescent of the Near-East for about 10,000 years ago, however, recent studies, based on RBP2 gene, confirmed that it was also grown on the Tibetan plateau in a similar era (Badr et al. 2000; Zohary and Hopf 2000; Wang et al. 2016a, b). Globally, barley is the fourth most important cereal crop grown after rice, wheat, and maize in terms of area and production (FAOSTAT 2017, https://www.fao.org/faostat:visited Oct. 2019). In 2016, barley production was reported more than 141 million tonnes globally with countries such as Ukraine, Canada, France, Germany, Australia, and Russia were the top producers (FAOSTAT 2017). Barley is mainly used as animal feed, human food, industrial raw materials for malting and brewing, and as an important ingredient in various healthy food recipes (Muñoz-Amatriaín et al. 2014b). At global scale, several biotic and abiotic factors are attributed to the lower barley productivity. Among abiotic stresses, heat stress, plays a major role in limiting barley yield. In barley, heat and drought stresses cause significant impact on both grain yield and malt qualities (Savin and Nicolas 1996; Macnicol et al. 1993). Mahalingam and Bregitzer (2019) reported that heat and drought stresses have greater impacts on grain yield and malting qualities, when barley is exposed to these stresses at reproductive than at vegetative stages. In India, barley grown in north western plains, including the states of Uttar Pradesh, Rajasthan, Haryana and Punjab, is exposed to terminal heat stress which causes significant losses in grain quality in addition to the yield (Verma et al. 2005). The issue becomes more important because this is the region from where the produce is mainly utilized for malting, while from other regions the crop is mainly utilised for feed purposes. In Indian sub-continent (Bangladesh, India, Nepal, and Pakistan) temperature rises suddenly during March and April as western hot wind exposes the reproductive phase of cereals including wheat and barley, to heat stress leading to loss in grain yield and quality.
Heat stress is generally reflected by reducing vegetative phase and poor grain development, making the grains unfit for malting and other uses. It causes early senescence in plants and a reduction in plant growth and yield along with variation in grain filling activities (Paulsen 1994). In addition, several physiological changes occur under heat stress, such as decline in photosynthesis, reproductive abortion, and reduced grain filling that results in reduced kernel weight (Wardlaw and Wrigley 1994). During grain filling in barley, rapid respiration occurs under heat stress at night, thereby further reducing the efficiency of the photosynthesis to fill the grain (Gent and Kiyomoto 1985; McCullough and Hunt 1989). Likewise, heat stress restricts the growing seasons and adversely affects the adaptability of the crop especially at the flowering time specifically during March–April in Indian sub-continent. As early maturity helps the crop to avoid the period of stress, the development of short duration cultivars is the most efficient strategy for minimizing yield loss from terminal heat stress in low-value cropping systems (Hegde 2010). However recent study indicates that the adequate timing of plant development is also crucial to maximize yield in barley under stress conditions that is related to temperature (Wiegmann et al. 2019). Sensitivity to combined stress was generally greater than heat or drought individually, and greater when imposed at heading than at the vegetative stages and the seeds from plants stressed during heading showed differences in malt extract, beta-glucan content and percent soluble protein (Mahalingam and Bregitzer, 2019). However, since it is mostly the escape mechanism, genetic incorporation of heat tolerance is important to get stable yield and grain development during stress conditions. Therefore, all stages of plant growth should be considered to assist breeding for heat tolerance in barley (Sallam et al. 2018).
Genetic improvement of crops depends on available variations in the germplasm which can be explored by a specific or a combination of methods. The vulnerability of crops increases towards biotic and abiotic stress due to the reduction of genetic diversity in given gene pool (Sharma et al. 2017). Therefore, plant breeding programs must incorporate new sources of genetic diversity in order to achieve higher levels of tolerances to biotic and abiotic stresses for increased yield. Molecular markers can be used for studying the genetic diversity among available genetic resources with breeders. Of these markers, simple sequence repeats (SSR) markers are well known to be used for such studies (Sharma et al. 2017; Sallam et al. 2018). In recent years, several high throughput customized SNP genotyping arrays have become available for a variety of crops including barley. The recently developed barley 50 K iSelect Illumina SNP platform provides a large number of SNP variants based on exome capture (Bayer et al. 2017). These SNPs can be used for genetic characterization of barley germplasm collection, QTL mapping, genome wide association studies (GWAS), marker assisted and genomic selection.
The combined information gained from genetic diversity and agro-morphological traits under heat stress condition can be valuable for improving heat tolerance in barley. A few recent studies used such an approach based on SSR (Sallam et al. 2018) and SNP (Amezrou et al. 2018a, b) markers in barley, where number of markers used were limited with a poor resolution of the barley genome. To the best of our knowledge, no such study has been done using 50 K iSelect barley SNP array for heat tolerance. Therefore, the current study was taken up to understand the agro-morphological and genetic diversity of a global association mapping panel 2017 (AM2017) based on high density SNP markers and to further determine the population structure with focus on heat tolerance and grain yield.
Materials and methods
Plant material
A set designated as Association Mapping Panel 2017 (AM2017) of 316 genotypes of spring barley, consisting of landraces and released cultivars from different countries over the years, and advanced breeding lines from the International Center for Agriculture Research in the Dry Areas (ICARDA), was used for this study. The AM2017 has an appropriate representation of two-row (173) and six-row (143) type barley genotypes across feed and malting types originating from different continents. Further, AM2017 can be characterized as 134 breeding lines from ICARDA’s barley breeding programs, 161 cultivars (genotype registered as cultivars in multiple countries across globe), and 21 landraces collected from different sources. The details of the genotypes are provided in the Supplementary Table S1.
DNA isolation and SNP genotyping
Genomic DNA was isolated from lyophilized young leaf tissue from a single plant at the seedling stage (Growth Stage 12) (Zadoks et al. 1974) as described in Slotta et al. (2008) at the Cereal Crop Research Unit, USDA-ARS, Fargo, North Dakota, USA. Genotyping was done using Illumina iSelect 50 K SNP array for barley (Illumina, San Diego, CA, USA) (Bayer et al. 2017) using the manufacturer’s guidelines. The iSelect 50 K SNP array comprises of 49,267 mapped loci (https://ics.hutton.ac.uk/50k/). Quality filtration of SNP markers was done using PLINK v1.7 (Purcell et al. 2007). All monomorphic markers were removed prior to the application of further filtration. Markers with minor allele frequency (MAF) of less than 5% and missing values of more than 20% were not considered for this study. Finally, only the markers with available genetic and physical map information associated with SNPs on 50 K array were kept (Bayer et al. 2017). Distribution of filtered SNP markers based on the physical map across the genome is illustrated using the CMplot R package (https://github.com/YinLiLin/R-CMplot).
Field experiment and phenotypic diversity
The AM2017 was assessed for heat stress tolerance in experiments conducted at the Research Farm (29°09′07.6″N 75°42′05.7″E) of the Department of Genetics and Plant Breeding, Chaudhary Charan Singh Haryana Agricultural University (CCSHAU), Hisar, India, for two successive crop seasons i.e., the year 2017–18 (November to March) and 2018–19 (December to April) (hereafter referred to as 2018 and 2019). The experiment was conducted in an augmented block design with genotype ‘BH946’ (latest released barley cultivar for north western plains of India) planted as a check (control) at the interval of 20 test entries. Each plot consisted of two rows of 2 m length with 0.3 m row spacing. Two sowing dates were followed each year viz. timely-sown (TS) (mid-November) and late-sown (LS) (mid-December) conditions to coincide the effect of heat stress in late sown crop over timely sown.
The phenological data were recorded on five randomly selected competitive plants from each genotype in each plot under timely and late sown experiments according to the barley descriptor (IPGRI 1994). These plants were tagged before heading and observations were recorded for plant height (PH), spike length (SL), and spikelet number per spike (SPK). Days to heading (DH) at 50% ear emergence and days to maturity (DM) at physiological maturity, grain yield per plot (GY/P) were recorded on the plot basis. Productive tillers per meter (T/M) and hectoliter weight (HW) were also recorded for each plot. Principal component analysis (PCA) was performed using ‘prcomp’ function in R statistical software (R Development Core Team 2018). All the traits were considered for the PCA. A comparative study between two sowing conditions was performed based on Ward’s minimum variance (Ward 1963) hierarchical clustering using DARwin v6.0.15 (Perrier, X. and Jacquemoud-Collet 2006). Pearson’s correlation coefficients were estimated between agro-morphological traits among and within sowing conditions using a corrplot R package (Wei and Simko 2017).
Genetic diversity and population structure analysis
The gene diversity (GD) and polymorphic information content (PIC) were estimated using POWERMARKER v3.25 (Liu and Muse 2005) on the filtered SNP markers. Based on the estimated PIC, 2,065 SNP markers bearing high PIC values representing a unique genetic map position throughout seven barley chromosomes were selected as highly informative markers for population structure analysis. StrAuto software tool (Chhatre and Emerson 2017) was used to run STRUCTURE 2.3.4 (Pritchard et al. 2000) in a Linux environment. The K values were tested for 1 to 10 with five independent runs for each K with 100,000 burn-in and 100,000 Monte Carlo Markov Chain (MCMC) iterations. The best K value was estimated using the web-based STRUCTURE HARVESTER tool (Earl and VonHoldt 2011). A Nei distance matrix (Nei 1972) was computed in POWERMARKER v3.25 using 2,065 SNPs and used as input for the phylogenetic analysis based on the Unweighted Pair-Group Method using Arithmetic mean (UPGMA) algorithm. The MEGA v7 (Kumar et al. 2016) was used to view the resulting dendrogram. Principal components analysis (PCA) was done in filtered SNP markers using ‘prcomp’ function in the R statistical program (R Development Core Team 2018). The extent of kinship among individuals was also estimated with the filtered set of SNP markers in GAPIT (Lipka et al. 2012) in R software (R Development Core Team 2018), using VanRaden method (VanRaden 2008).
Results
SNP marker
The current study panel was genotyped using the 50 K Illumina iSelect SNP array comprised of more than 49 K SNP markers. After removing the monomorphic markers, 39,960 markers were contained. Markers removed upon quality filtration based on MAF and missingness test resulted in 36,856 markers. Further, 60 markers were removed lacking physical map position information and 3 markers lacking chromosomal information. None of the genotypes failed the missingness test due to low genotyping. Finally, 36,793 quality SNP markers were filtered for 316 genotypes and used in further genetic analyses. The distribution of 36,793 markers is illustrated in Fig. 1 in 1 Mb window size. The maximum number of markers was observed in chromosome 5H (18.41%) followed by 2H (16.91%). The chromosome 1H had the lowest number (10.84%) of SNPs amongst the seven chromosomes. The total genetic distance covered by the filtered markers was 991.82 cM (Table 1). An average inter-marker distance of 0.0270 cM/SNP and/or average marker density of 37.09 SNPs/cM was observed in this study.

Distribution of SNP markers obtained post quality filtration in the barley genome based on the physical map. Color legend on the right shows the marker density
SNP based genetic diversity
Genetic diversity was estimated based on the filtered set of 36,793 SNPs. The gene diversity (GD) ranged from 0.1 (2,712 SNPs) to 0.5 (12,882 SNPs). Maximum GD was observed in chromosome 3H (0.369) and the minimum in chromosome 4H (0.355) with an overall average of 0.362 in the barley genome. An average PIC of 0.2893 was found in AM2017 which ranged from 0.1 (3,466 SNPs) to 0.4 (12,595 SNPs). The chromosome 3H and 4H had the highest (0.2935) and lowest (0.2850) PIC, respectively (Table 1). The average minor allele frequency (MAF) was 0.2729.
Population structure
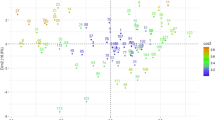
Based on the Bayesian model, population structure was estimated using highly informative selected markers (2,065 SNPs) in 316 genotypes of AM2017. The plot between delta K (∆K) and the number of K showed a sharp peak at K = 2 (Fig. 2a). Subpopulation 1 had 40.18% of total genotypes, whereas subpopulation 2 had 59.81% (Fig. 2c). It was observed that subpopulation 1 had 97.63% of two-row barley genotypes with only 3 genotypes of 6-row type. Subpopulation 2 had 74.07% of six-row barley genotypes with remaining genotypes to be two-row in nature (Supplementary Table S1). The highly informative SNPs used for generating a UPGMA based phylogenetic tree (Fig. 5b), concurred with the results of population structure i.e., two distinct clusters. A small peak can be seen at K = 4 (Fig. 2a), which upon careful observation divided each of the two subpopulations into two sub-groups (i.e. 1A; 1B and 2A; 2B) (Fig. 2d). The kinship analysis also corroborated the presence of two main genetic clusters (Fig. 5b) based on the row types of barley genotypes. Principal component analysis (PCA) based on the filtered set of 36,793 SNP markers also supported the presence of two subpopulations in the AM2017. Two clusters were observed in the PCA, where principal components (PCs) 1 and 2 explained 9.60% and 4.13% of the total variation, respectively. Subpopulation 1 and 2 can be observed as separate clusters in the PCA scatter plot (Fig. 3).

Population structure and kinship analysis; a line plot between delta-K and number of possible clusters (K). The highest peak at K = 2 supports the presence of two subpopulations in the AM2017; b Heat map of kinship matrix with a distance-based hierarchical clustering of the 316 barley genotypes calculated from the filtered set of 36,793 SNP markers. The heat map also represents a low level of relatedness among the genotypes of AM2017; c, and d are bar plots representing STRUCTURE clustering results obtained at K = 2 and K = 4. Each barley genotype is aligned as per the order in the kinship matrix and represented by a thin bar corresponding to the sum of assigned probabilities to the K clusters

Principal component analysis (PCA) based on 36,793 SNP markers in the AM2017 panel. The PC1 and PC2 represents 9.60 and 4.13% of the total variation. The row-types and subpopulations are represented using different legends
Agro-morphological traits
The mean values of eight agro-morphological traits measured for AM2017 in timely sown (TS) and late sown (LS) conditions are presented in Table 2. The genotype wise agro-morphological traits are presented in Supplemental Table S2. In TS, days to heading ranged from 75 to125 days with an average of 99 days. In LS, it ranged from 65 to 101 days with an average of 87 days. Early maturity was observed in LS with a range of 98 to 137 days over TS where maturity ranged from 107 to 147 days. The difference in average days to maturity was 15 days between two sowing conditions. Effect of heat stress was observed in the form of reduced performances of genotypes in LS compared to the TS. Reduced plant height of AM2017 genotypes was observed in LS condition compared to the TS. Average plant height in TS (PH-TS) was 100 cm (range 72–129 cm) whereas in LS, plant height was found 80 cm (range 60–110 cm). A similar pattern of reduction in performances of barley genotypes for other traits such as tillers per meter, grain yield per plot, spike length, spikelets per spike and hectoliter weight was observed in LS compared to the TS (Table 2, Supplemental Table S2). For plant height (PH), 20% reduction in height was observed in LS compared to the TS. The highest reduction in performance of trait was observed for T/M (36.92%) followed by GY/P (21.28%). A similar reduction pattern in the performance of other traits was observed such as DH (11%), DM (12%), SL (4.9%) and SPK (8.8%), due to the effect of heat stress in LS. In hectoliter weight, only 4.1% reduction was observed in LS compared to the TS. The summary statistics of all agro-morphological traits are presented in Table 2.
Phenotypic diversity based on agro-morphological traits
Using 16 quantitative traits from TS and LS, the first three Principal Components (PCs) accounted for 55.6% of the total variation in the PCA. The PC1 and PC2 explained 24.6% and 19.7% of the total variability, respectively (Fig. 4). In the first PC, variables DH and DM in LS followed by DH and DM in TS had high positive loadings. The PH in TS had the largest negative loadings in PC1. In PC2, PH-TS and GY/P-LS had the highest positive loadings while SPK and SL in both TS and LS had the largest negative loadings. The PC3 (11.20%) was associated DH and DM in TS with high positive loadings and GY/P in both TS and LS with largest negative loadings (Table 2). Based on the performance of agro-morphological traits, the hierarchical clustering, after 1,000 bootstraps, genotypes were grouped into six clusters. Most of the genotypes from LS and TS were observed as separate clusters (Fig. 5a). Few genotypes clustered with similar performance when compared with these two conditions. Due to agglomerative dissimilarity, seven clusters can be considered, where each cluster was observed to be dominated by genotypes from either TS or LS condition except for clusters V and VI (Fig. 5a). Clusters I, II, and IV were mainly comprised of TS genotypes, whereas clusters III and VII of LS genotypes. Some genotypes were observed as exceptions to such classification. It indicated earlier, genotypes in LS condition showed reduced performance considering the traits under study, genotypes from LS condition falling under TS dominated cluster can be considered as heat stress tolerant under stressed condition (Supplemental Table S2).

Principal component analysis (PCA) based on genetic distance computed for 316 barley genotypes in timely-sown condition (TS) and late-sown condition (LS) by using 16 agro-morphological traits

a Phenotypic diversity due to agro-morphological traits: Hierarchical clustering based on the observations taken for 8 agro-morphological traits on 316 genotypes from TS and LS. Separate colors are used to distinguish observations from TS (green) and LS (red). b Genetic diversity: Highly informative SNP markers (2065) depicting two major clusters in the UPGMA based phylogenetic tree. The two clusters obtained concurred with results of population structure of having two subpopulations in the panel. The subpopulations are represented with separate colors
Correlation between phenotypic traits
In TS condition, positive significant correlations were observed between DH-DM, DH-T/M, DM-T/M, T/M-GY/P, SL-SPK, HW-T/M, and HW-SPK at p < 0.05. The negative correlations were observed between DH-PH, DM-PH, PH-T/M, SL-GY/P (Fig. 6 and Supplemental Table S3). In LS condition, a strong correlation was observed between DH and DM (r2=0.9) as compared to that in the TS condition (r2 = 0.8). At a significance level p < 0.001, the only negative correlation was observed between GY/P and SL. DH and DM were found positively correlated with T/M, GY/P and HW whereas T/M and GY/P were also positively correlated. There was a positive correlation among SL, SPK, and HW while HW had positive correlation GY/P. At a significance level p < 0.01, negative and positive correlations were found between DM-SL and T/M-HW, respectively (Fig. 6, Supplemental Table S3).

A Pearson’s correlation coefficient (r2) plot between 16 traits in the AM2017 panel measured in timely-sown (TS) and late-sown (LS) conditions. The color legend shows the magnitude of the correlation coefficient between any given pair of traits. Significance levels at probability (p) < 0.001, < 0.01, and < 0.05 are denoted with ***, **, and *, respectively
Discussion
Phenotypic diversity.
In the current study, the phenotypic and molecular diversity of 316 barley genotypes representing the landraces, cultivars from Asia, Africa, Europe, America, and advanced breeding lines from spring barley breeding program of ICARDA across high and low input conditions is reported. The spring barley breeding program of ICARDA includes genotypes selected for food, feed and malting purposes, intended for favorable as well as restrictive environment which is different than the genetic diversity report of Amezrou et al. (2018a) as it was confined to low input barley breeding program of ICARDA. The research work of Amezrou et al. (2018a) has already contributed to several agronomic traits, including GWAS of complex traits such as disease resistance and nutrient uptake (Amezrou et al. 2018a, b; Gyawali et al., 2020, 2019a, 2019b, 2018, 2017a, 2017b) Genetic variation is a pre-requisite and crucial for successful barley improvement for agronomic and quality traits (Muñoz-Amatriaín, 2014a; Amezrou et al. 2018a). Recently, Monteagudo et al. (2019) showed significant barley improvement in several agronomic traits including drought tolerance in Mediterranean environments by selecting genotypes from a diverse Spanish barley core collection. In the current study, we have shown higher genetic diversity in AM2017 panel of the underlying agronomic traits under normal and heat stressed conditions. Considering a diverse origin of AM2017, high agronomic and molecular diversity of the barley genotypes were expected in this panel. The principal component analysis of agro-morphological traits showed days to heading, days to maturity, spike length, spikelet number, productive tiller/meter, hectoliter weight, and yield/plot contributed to variations in AM2017 under timely and late sown conditions. These traits have potential for making genetic gain to improve heat tolerance in barley. We found that traits like spike length, spikelet number per spike, hectoliter weight and yield per plot are major yield components in AM2017 and potential targets for improving yield under TS and LS conditions in India. The LS conditions in North Western Plains Zone of India expose barley to the hot wind during the grain filling period till maturity, thereby adversely affecting grain filling process. As a result, the heat stress condition in cereals, including barley results into shriveled grains leading to yield losses as well as making grains unfit for end use specifically malting and other industrial uses. It is evident that grain weight contributes to yield under favorable conditions prevailing during the period of grain filling. In two-row barley, the reduced number of grains per spikes is one of the important yield components adversely affecting the grain yield in comparison to six row barley, but thousand kernel weight (TKW) and higher tillers per unit area compensates the lesser number of grains per spike (Evans and Wardlaw 1976; Wiebe 1979). In dry areas, moisture and heat stresses are frequently observed at all stages, especially grain filling, and ICARDA barley breeders tend to select material based on grain weight and other yield components for their target environments.
The correlation analyses of agro-morphological traits revealed that the yield and yield components had higher correlation under normal and late sown conditions. This has implication in selection of barley for stress conditions such as drought and heat stresses. Our study, using AM2017, showed that DH and DM had significant correlations with yield/plot in TS and LS conditions. In cereals breeding, a trade-off of key traits should be taken into consideration during selection and improving grain yield and quality. Both positive and negative correlations were found between key traits (Amezrou et al. 2018a) of low input barley breeding program which is in consistence with our studies and the findings from previous studies on agronomic traits of barley. Monteagudo et al. (2019) reported both positive and negative correlations among phenological traits and yield components. The introductions of novel alleles from diverse collections into breeding population have implication on selection strategies of these traits under normal and stress conditions. Therefore, in AM2017, a selection strategy of early to medium maturing barley genotypes with higher grain yield will result faster grain filling and increased SPK thereby, reducing the effects of heat stress under late sown condition.
Genetic diversity.
The current study is one of the few studies where a specific population (AM2017) was deliberately assembled and analyzed, representing very diverse barley genotypes consisting of advanced lines of ICARDA spring barley breeding program, cultivars and landraces from different parts of the globe to provide a platform of GWAS for several important traits. The genetic diversity study of AM2014 reported by Amezrou et al. (2018a) provided a platform for several GWAS studies such as foliar diseases (NFNB, SFNB, spot blotch) and multiple element contents (Amezrou et al. 2018a, b; Gyawali et al. 2018, 2017b). For genotyping of AM2017, we used the 50 K SNP markers, which offer a highly polymorphic, co-dominant, accurate, and high-throughput marker system. SNP markers can be used in germplasm characterization and selection of desirable alleles in breeding programs using markers assisted selection (Lombardi et al. 2014; Amezrou et al. 2018a). In our study, the average gene diversity was 0.362, which is similar to the values reported by Amezrou et al. (2018a), while, genetic diversity of AM2017 was slightly higher than previous studies reported by Sun et al. (2010) and Rodriguez el al. (2012) using SSR markers, 0.338 and 0.298, respectively.
Higher SNP diversity in the AM2017 mapping panel was expected because of inclusion of diverse genotypes, originated from different barley breeding programs across globe, representing feed, food, and malting barley for rainfed or irrigated production conditions. Varshney et al. (2010) reported higher genetic diversity (SNP and SSR) of ICARDA barley germplasm collection due to inclusion of genotypes from diverse origin and geographic region as well as introgression from landraces and H. spontaneum. They suggested that ICARDA barley germplasm was suitable for making higher genetic gain on abiotic stress tolerances, specifically drought and heat stress tolerances. Selecting SNPs based on their high polymorphism levels is desirable for mapping and marker-based selection strategies (Jones et al. 2007). The average distance per SNP of AM2017 ranged from 0.025–0.033 cM among seven barley chromosomes with an average distance of 0.027 cM per SNP. The marker density of AM2017 was much higher than average marker density of 0.231 cM of AM2014 as reported by Amezrou et al. (2018a). A marker density of 1 DArT marker per 1.5 cM (Comadran et al. 2009) and 1 SNP marker per 0.72 cM (Pasam et al. 2012; Cockram et al. 2010; Massman et al. 2011) was reported in earlier barley mapping populations. The GWAS have been successfully carried out using much lower marker density in earlier mapping panels/populations compared to the current study (Comadran et al. 2012, Muñoz-Amatriaín et al. 2014b, Tamang et al. 2015, Amezrou et al. 2018a, b, Gyawali et al. 2017b; Gyawali et al. 2018). The 50 k SNP platform of AM2017 gave a higher SNP density making this panel suitable for GWAS and genomic studies in future. The distribution of SNP markers in seven barley chromosomes (Fig. 1) suggest that SNP markers were well distributed across all chromosomes. The uniform distribution of SNP markers is one of the pre-requisites for any genetic and genomic studies. Therefore, we suggest that current average distance of SNP (0.027) and uniform distribution of SNP markers are highly desirable for GWAS and genomic studies using AM2017.
Population structure
Structure and principal component analyses of AM2017, with few exceptions, separated barley genotypes into two major subgroups, each representing two-row and six-row types. The results of AM2017 are in consistence with previous studies showing two-row and six-row sub-populations (Lasa and Igartua 2001; Chaabane et al. 2009; Chen et al. 2014; Usubaliev et al. 2013; Amezrou et al. 2018a). In barley, number of fertile florets is controlled by two major genes, vrs1 in 2H and int-c in 4H (Franckowiak and Lundqvist 1997; Cuesta-Marcos et al. 2010; Ramsay et al. 2011; Gyawali et al. 2017b). In AM2014, Gyawali et al. (2017b) mapped3 SNPs, 11_20606, 11_21070, and 11_20422, as significantly (q < 0.05) correlated with row types (fertility of lateral spikelets), including three other SNPs in the 4HS chromosome at the span of 38.44–53.87 cM. The further investigation of two sub-populations in AM2017 indicated that 2_row_Sub-pop_1 and 6_row_Sub-Pop_2 were distinct from each other while genotypes of 2_row_Sub-Pop_2 and 6_row_Sub_Pop_1 were overlapping each other (Fig. 3). Historically, in the spring barley breeding programs of ICARDA, two-row by six-row hybridization is frequently practiced (Amezrou et al. 2018a). Likewise, the AM2017 included cultivars from India, where similar hybridization is practiced for improving the adaptability of exotic two-row barleys with indigenous six-row cultivars (Verma et al. 2010). Therefore, the overlap between 2_row_Sub-Pop_2 and 6_row_Sub_Pop_1 might be as a result of historic two-row by six-row crosses in the barley breeding programs of ICARDA, India and other places. In AM2017, for detecting the most confident QTLs and avoid spurious associations, it is important to consider association mapping independently in the two sub populations specifically two-row and six-row types subpopulations. The SNP genotyping of AM2017 with 50 K iSelect Illumina SNP platform provides large number of SNP variants for both two-row and six-row subpopulations. These SNPs can be used for genetic characterization of germplasm collection and entire genome coverage with high resolution and will make the diversity contained in it efficiently accessible to barley breeders.
Conclusions
The agro-morphological and genetic studies of AM2017 provided a detailed description of genetic diversity and population structure of barley genotypes. It is evident that AM2017 is highly diverse in relation to many agronomic traits under timely (normal) and late sown (heat stress) conditions in India and neighboring countries. Agronomic traits including DH, DM, SKP, SL, and G/P were important for selecting heat tolerance in barley. We have demonstrated that a strong population structure exists in AM2017. Therefore, we recommend dissection of population structure of AM2017, based on row types, and will be useful in future GWAS to avoid any spurious associations. The recently developed 50 K iSelect Illumina SNP platform provides large number of SNP variants in barley genotypes. The high density of SNP markers in AM2017 shall provide a platform for successful GWAS and genomic studies of important traits for feed, food, and malting barley. The unprecedented level of information generated in the study will be a significant addition to barley genetics and breeding in future.
References
Amezrou R, Gyawali S, Belqadi L, Chao S, Arbaoui M, Mamidi S et al (2018) Molecular and phenotypic diversity of ICARDA spring barley (Hordeum vulgare L.) collection. Genet Resour Crop Evol 65:255–269. https://doi.org/10.1007/s10722-017-0527-z
Amezrou R, Verma RPS, Chao S, Brueggeman RS, Belqadi L, Arbaoui M, Rehman S, Gyawali S (2018) Genome-wide association studies of net form of net blotch resistance at seedling and adult plant stages in spring barley collection. Mole Breed 38:58. https://doi.org/10.1007/s11032-018-0813-2
Badr A, Müller K, Schäfer-Pregl R, El Rabey H, Effgen S, Ibrahim HH et al (2000) On the origin and domestication history of barley (Hordeum vulgare). Mol Biol Evo 17(4):499–510. https://doi.org/10.1093/oxfordjournals.molbev.a026330
Bayer MM, Rapazote-Flores P, Ganal M, Hedley PE, Macaulay M, Plieske J et al (2017) Development and evaluation of a barley 50k iSelect SNP array. Front Plant Sci 8:1792. https://doi.org/10.3389/fpls.2017.01792
Chaabane R, El-Felah M, Salah HB, Abdelly C, Ramla D, Nada A, Saker M (2009) Molecular characterization of Tunisian barley (Hordeum vulgare L.) genotypes using microsatellites (SSRs) markers. Eur J Sci Res 36(1):6–15
Chen H, Xie W, He H, Yu H, Chen W, Li J et al (2014) A high-density snp genotyping array for rice biology and molecular breeding. Mol Plant 7:541–553. https://doi.org/10.1093/mp/sst135
Chhatre VE, Emerson KJ (2017) StrAuto: automation and parallelization of STRUCTURE analysis. BMC Bioinformatics 18:192. https://doi.org/10.1186/s12859-017-1593-0
Cockram J, White J, Zuluaga LD, Smith D, Comadran J, Macaulay M et al (2010) Genome-wide association mapping to candidate polymorphism resolution in the unsequenced barley genome. PNAS 107(50):21611–21616. https://doi.org/10.1073/pnas.1010179107
Comadran J, Thomas WT, van Eeuwijk FA, Ceccarelli S, Grando S, Stanca AM, Pecchioni N, Akar T, Al-Yassin A, Benbelkacem A, Ouabbou H, Bort J, Romagosa I, Hackett CA, Russell JR (2009) Patterns of genetic diversity and linkage disequilibrium in a highly structured Hordeum vulgare association-mapping population for the Mediter-ranean basin. Theor Appl Genet 119:175–187. https://doi.org/10.1007/s00122-009-1027-0
Comadran J, Kilian B, Russell J, Ramsay L, Stein N, Ganal M et al (2012) Natural variation in a homolog of Antirrhinum CENTRORADIALIS contributed to spring growth habit and environmental adaptation in cultivated barley. Nat Genet 12:1388–1392. https://doi.org/10.1038/ng.2447
Cuesta-Marcos A, Szucs P, Close TJ, Filichkin T, Muehlbauer GJ, Smith KP, Hayes PM (2010) Genome-wide SNPs and re-sequencing of growth habit and inflorescence genes in barley: implications for association mapping in germplasm arrays varying in size and structure. BMC Genom 11:707
Earl D, VonHoldt B (2011) STRUCTURE HARVESTER: a websiteand a program for vizualizing STRUCTURE output nad implementing the Evanno method. Conserv Genet Resour 4(2):359–361
FAOSTAT (2017) Crops/Regions/World List/Production Quantity for Barley. Available at https://www.fao.org/faostat/en/#data/QC (accessed on August 2019)
Franckowiak JD, Lundqvist U (1997) Barley genetics symposium 6, six-rowed spike 1, vrs1. Barley Genet Newsl 26:49–50
Gent MPN, Kiyomoto RK (1985) Comparison of canopy and flag leaf net carbon dioxide exchange of 1920 and 1977 New York winter wheats. Crop Sci 25(1):81–86. https://doi.org/10.2135/cropsci1985.0011183x002500010021x
Gyawali S, Amezrou R, Verma RPS, Brueggeman RS, Rehman S, Belqadi L, Arbaoui M, Tamang P, Singh M (2020) Seedling and adult stage resistance to net form of net blotch (NFNB) in spring barley and stability of adult stage resistance to NFNB in Morocco. J Phytopathol 168(5):254–266. https://doi.org/10.1111/jph.12887
Gyawali S, Amezrou R, Verma RPS, Brueggeman RS, Rehman S, Belqadi L, Arbaoui M, Tamang P, Singh M (2019) Seedling and adult stage resistance to spot form of net blotch (SFNB) in spring barley and stability of adult stage resistance to SFNB in Morocco. Eur J Plant Pathol 153:475–487. https://doi.org/10.1007/s10658-018-1575-8
Gyawali S, Otte ML, Jacob DL, Abderrazek J, Verma RPS (2019) Multiple element concentration in the grain of spring barley (Hordeum vulgare L.) collection. J Plant Nutri 12(9):1036–1046. https://doi.org/10.1080/01904167.2019.1589507
Gyawali S, Chao S, Vaish SS, Singh SP, Rehman S, Vishwakarma SR, Verma RPS (2018) Genome wide association studies (GWAS) of spot blotch resistance at the seedling and the adult plant stages in a collection of spring barley. Mole Breed 38:62. https://doi.org/10.1007/s11032018-0815-0
Gyawali S, Verma RPS, Kumar S, Bhardwaj SC, Gangwar OP, Selvakumar R, Shekhawat PS, Rehman S, Sharma-Poudyal D (2017) Seedling and adult-plant stage resistance of a world collection of barley genotypes to stripe rust. J Phytopathol 166(1):18–27. https://doi.org/10.1111/jph.12655
Gyawali S, Otte ML, Chao S, Jilal A, Jacob DL, Amezrou R, Verma RPS (2017) Genome wide association studies (GWAS) of element contents in grain with a special focus on zinc and iron in a world collection of barley (Hordeum vulgare L.). J Cereal Sci 77:266–274. https://doi.org/10.1016/j.jcs.2017.08.019
Hegde VS (2010) Genetics of flowering time in chickpea in a semi-arid environment. Plant Breed 129:683–687. https://doi.org/10.1111/j.1439-0523.2009.01748.x
IPGRI (1994) Descriptors for barley (Hordeum vulgare L.). International Plant Genetic Resources Institute, Rome Italy: https://www.bioversityinternational.org/e-library/publications/detail/descriptors-for-barley-hordeum-vulgare-l/. (accessed September 2017)
Kumar S, Stecher G, Tamura K (2016) MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874. https://doi.org/10.1093/molbev/msw054
Lasa J, Igartua E (2001) Morphological and agronomical diversity patterns in the Spanish barley core collection. Hereditas 135(23):217–225
Lipka AE, Tian F, Wang Q, Peiffer J, Li M, Bradbury PJ et al (2012) GAPIT: Genome association and prediction integrated tool. Bioinformatics 28:2397–2399. https://doi.org/10.1093/bioinformatics/bts444
Liu K, Muse SV (2005) Power Marker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21:2128–2129
Lombardi M, Materne M, Cogan NOI, Rodda M, Daetwyler HD, Slater AT, Forster JW, Kaur S (2014) Assessment of genetic variation within a global collection of lentil (Lens culinaris Medik.) cultivars and landraces using SNP markers. BMC Genet 15:150. https://doi.org/10.1186/s12863-014-0150-3
Macnicol PK, Jacobsen JV, Keys MM, Stuart IM (1993) Effects of heat and water-stress on malt quality and grain parameters of Schooner barley grown in cabinets. J Cereal Sci 18:61–68
Mahalingam R, Bregitzer P (2019) Impact on physiology and malting quality of barley exposed to heat, drought and their combination during different growth stages under controlled environment. Physiol Plant 165:277–289. https://doi.org/10.1111/ppl.12841
McCullough DE, Hunt LA (1989) Respiration and dry matter accumulation around the time of anthesis in field stands of winter wheat (Triticum aestivum). Ann Bot 63:321–329. https://doi.org/10.1093/oxfordjournals.aob.a087748
Monteagudo A, Casas AM, Cantalapiedra CP, Contreras-Moreira B, Gracia MP, Igartua E (2019) Harnessing novel diversity from landraces to improve an elite barley variety. Front Plant Sci 10:434. https://doi.org/10.3389/fpls.2019.00434
Muñoz-Amatriaín M, Cuesta-Marcos A, Hayes PM, Muehlbauer GJ (2014a) Barley genetic variation: implications for crop improvement. Brief Funct Genomics 13(4):341–350. https://doi.org/10.1093/bfgp/elu006
Munoz-Amatriaın M, Cuesta-Marcos A, Endelman JB, Comadran J, Bonman JM, Bockelman HE, Chao S, Russel J, Waugh R, Hayes PM, Muehlbauer GJ (2014b) The USDA barley core collection: genetic diversity, population structure, and potential for genome-wide association studies. PLoS ONE 9(4):e94688. https://doi.org/10.1371/journal.pone.0094688
Massman J, Cooper B, Horsley R, Neate SM, Dill-Macky R, Chao S, Dong Y, Schwarz P, Muehlbauer GJ, Smith KP (2011) Genome-wide association mapping of Fusarium head blight resistance in contemporary barley breeding germplasm. Mol Breed 27:439–454
Nei M (1972) Genetic distance between populations. Am Nat 106:283–292
Pasam RK, Sharma R, Malosetti M, van Eeuwijk FA, Haseneyer G, Kilian B, Graner A (2012) Genome-wide association studies for agronomical traits in a worldwide spring barley collection. BMC Plant Biol 12:16. https://doi.org/10.1186/1471-2229-12-16
Paulsen GM (1994) High temperature responses of crop plants. In: Physiology and determination of crop yield. Eds: Boote KJ, Bennett JM, Sinclair TR, Paulsen GM. American Society of Agronomy, Inc Crop Science Society of America, Inc Soil Science Society of America, Inc. Pub: Wiley, NewYork. pp 365–389
Perrier X, Jacquemoud-Collet J (2006) DARwin Software. https://darwin.cirad.fr/darwin.
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959. https://doi.org/10.1111/j.1471-8286.2007.01758.x
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D et al (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81:559–575. https://doi.org/10.1086/519795
R Development Core Team (2018) R: A language and environment for statistical computing. Vienna, Austria. doi:R Foundation for Statistical Computing, Vienna, Austria. ISBN 3–900051–07–0, URL https://www.R-project.org.
Ramsay L, Comadran J, Druka A, Marshall DF, Thomas WT, Macaulay M, MacKenzie K, Simpson C, Fuller J, Bonar N (2011) INTERMEDIUM-C, a modifier of lateral spikelet fertility in barley, is an ortholog of the maize domestication gene TEOSINTE BRANCHED 1. Nat Genet 43:169–172
Rodriguez M, Rau D, Sullivan D, Brown AHD, Papa R, Attene G (2012) Genetic structure and linkage disequilibrium in landrace populations of barley in Sardinia. Theor Appl Genet 125:171–184. https://doi.org/10.1007/s00122-012-1824-8
Sallam A, Amro A, EL-Akhdar A, Dawood MFA, Kumamaru T, Stephen Baenziger P, (2018) Genetic diversity and genetic variation in morpho-physiological traits to improve heat tolerance in spring barley. Mol Biol Rep 45:2441–2453. https://doi.org/10.1007/s11033-018-4410-6
Savin R, Nicolas ME (1996) Effects of short periods of drought and high temperature on grain growth and starch accumulation of two malting barley cultivars. Aust J Plant Physiol 23:201–210
Sharma P, Sareen S, Saini M, Shefali, (2017) Assessing genetic variation for heat stress tolerance in Indian bread wheat genotypes using morpho-physiological traits and molecular markers. Plant Genet Resour 15(6):539–547. https://doi.org/10.1017/S1479262116000241
Slotta TAB, Brady L, Chao S (2008) High throughput tissue preparation for large-scale genotyping experiments. Mol Ecol Resour 8:83–87. https://doi.org/10.1111/j.1471-8286.2007.01907.x
Sun D, Ren W, Sun G, Peng J (2010) Molecular diversity and association mapping of quantitative traits in Tibetan wild and worldwide originated barley (Hordeum vulgare L.) germplasm. Euphytica 178:31–43. https://doi.org/10.1007/s10681-010-0260-6
Tamang P, Neupane A, Mamidi S, Friesen T, Brueggeman R (2015) Association mapping of seedling resistance to spot form of net blotch in a worldwide collection of barley. Phytopathology 105:500–508
Usubaliev B, Brantestam AK, Salomon B, Garkava-Gustavson L, Bothmer R (2013) Genetic diversity in farmer grown spring barley material from Kyrgyzstan. Genet Resour Crop Evol 60:1843–1858. https://doi.org/10.1007/s10722-013-9959-2
VanRaden PM (2008) Efficient methods to compute genomic predictions. J Dairy Sci 91:4414–4423. https://doi.org/10.3168/jds.2007-0980
Varshney RK, Baum M, Guo P, Grando S, Ceccarelli S, Graner A (2010) Features of SNP and SSR diversity in a set of ICARDA barley germplasm collection. Mol Breeding 26:229–242. https://doi.org/10.1007/s11032-009-9373-9
Verma RPS, Sharma RK, Mishra B (2005) Future of barley for malt, feed and fodder in India. Directorate of Wheat Research, Karnal-132001, India. Technical Bulletin No.9: P28.
Verma RPS, Sarkar B, Mishra B (2010) Improvement of two-row malt barley through two x six-row hybridization in India. In: Proc. 10th International barley genetics symposium, 5–10, April, 2008. Alexandria, Egypt. Edi: Ceccarelli, S. and Grando, S. ICARDA, PO Box 5466, Aleppo, Syria. pp 432–442.
Verma RPS, Selvakumar R, Gangwar OP, Shekhawat PS, Bhardwaj SC, Rehman S, Sharma Poudyal D, Gyawali S (2018) Identification of additional sources of resistance to Puccinia striiformis f. sp. hordei (PSH) in a collection of barley genotypes adapted to the high input condition. J of Phytopathol 166(5):355–364. https://doi.org/10.1111/jph.12693
Wang J, Sun G, Ren X, Li C, Liu L, Wang Q et al (2016) QTL underlying some agronomic traits in barley detected by SNP markers. BMC Genet 17:103. https://doi.org/10.1186/s12863-016-0409-y
Wang Y, Ren X, Sun D, Sun G (2016) Molecular evidence of RNA polymerase II gene reveals the origin of worldwide cultivated barley. Sci Rep 6:36122. https://doi.org/10.1038/srep36122
Ward JH (1963) Hierarchical grouping to optimize an objective function. J Am Stat Assoc 58(301):236–244. https://doi.org/10.1080/01621459.1963.10500845
Wardlaw IF, Wrigley CW (1994) Heat tolerance in temperate cereals: an overview. Australian J of Plant Physiol 21(6):695–703. https://doi.org/10.1071/pp9940695
Wei T, Simko V (2017) R package “corrplot”: Visualization of a correlation matrix (Version 0.84). https//github.com/taiyun/corrplot. Available at: https://github.com/taiyun/corrplot.
Wiegmann M, Maurer A, Pham A, March TJ, Al-Abdallat A, Thomas WTB, Bull HJ, Shahid M, Eglinton J, Baum M, Flavell AJ, Tester M, Pillen K (2019) Barley yield formation under abiotic stress depends on the interplay between flowering time genes and environmental cues. Sci Reports 9:6397
Zadoks J, Chang T, Konzak C (1974) A decimal growth code for the growth stages of cereals. Weed Res 14:415–421
Zohary D, Hopf M (2000) Domestication of plants in the old world, 3rd edn. Press. Oxford, Univ. https://doi.org/10.1517/17425247.2013.856877.Intranasal
Acknowledgements
The first author sincerely acknowledges the financial support received from ICARDA for attending the GWAS training program at Morocco, which formed the basis for this manuscript as well as for providing the experimental material (association mapping population 2017 [AM2017]) for carrying out PhD research at CCSHAU, Hisar, India.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Verma, S., Yashveer, S., Rehman, S. et al. Genetic and Agro-morphological diversity in global barley (Hordeum vulgare L.) collection at ICARDA. Genet Resour Crop Evol 68, 1315–1330 (2021). https://doi.org/10.1007/s10722-020-01063-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10722-020-01063-7