
Abstract
Unraveling the cryptic genetic diversity and selective breeding network in various Porphyra strains is of significance for conservation and utilization of economically important nori crops, for both current and future needs. Here, we used nuclear ribosomal spacer (ITS1) region to investigate the genetic variation and intra-specific relatedness of 59 Porphyra yezoensis Ueda specimens worldwide using phylogenetic and parsimony genealogical approaches. 23 nrDNA ITS1 genotypes were revealed and clustering analysis grouped them into two distinct clades. High genetic diversity was detected in wild P. yezoensis strains from Miyagi and Hokkaido Prefectures in Japan, while the cultivated strains from China and South Korea exhibited relatively higher genetic diversity likewise, which provided crucial genetic insights for future commercial breeding of P. yezoensis on a global scale. In addition, phylogenetic study has revealed the genetic relationship of strains with unknown parentage to those with known parentage, and also ITS1 sequence pattern could be correlated with the geographic origin of P. yezoensis specimens. All these pedigree information generated from this research can be used to select parents for inter-specific or intra-specific selective breeding and cross breeding to maximize the preservation of stock resource and sustainable development of nori industry.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The red algal genus Porphyra C. Agardh (known commonly as nori in Japan, and Zicai in China) has a dimorphic life cycle with an alternation between a macroscopic foliose thallus (gametophytic phase) and a microscopic filamentous thallus (sporophytic phase). In this genus, cultivated P. yezoensis Ueda is one of the most economically important marine crops in East Asia (Niwa and Aruga 2003; Niwa et al. 2008). Since the 1960s with the development of synthetic fiber nets, strain-preservation and artificial seeding in Porphyra mariculture, nearly 60 strains of P. yezoensis have been isolated and used for cultivation in Japan, China and South Korea (Kunimoto et al. 1999; Iitsuka et al. 2002; Hwang et al. 2005; Weng et al. 2005; Hu et al. 2007; Niwa et al. 2008, 2009). Although these cultivars have considerably contributed to increasing yields, some subsequent problems occurred in nori farms. (1) Sometimes the origin of cultivars for mariculture is uncertain owing to the exchange of P. yezoensis cultivars with quite diverse genetic backgrounds among different growing regions in Asia, or the loss of originally historical maintenance records, which leave behind some hidden troubles for following selection and breeding. (2) It is extremely essential to keep the developed strains or wild strains absolutely pure for the germplasm conservation and advancement of Porphyra breeding, however, simple but highly variable morphological characteristics disenable nori breeders to classify their P. yezoensis cultivars with certainty, and nori cultivators even often blend several conchocelis strains when they seed nori nets with conchospores (Niwa et al. 2004). (3) Recent molecular investigations indicated that as the predominant selective breeding species, P. yezoensis has resulted in increased genetic uniformity in the stock resource for nori cultivation (Niwa et al. 2004; Niwa and Aruga 2006). However, the cultivated P. yezoensis strains have a lower genetic diversity than wild strains (Kunimoto et al. 1999), and then inevitably, the intensive selective breeding of cultivated P. yezoensis can lead to reduced genetic diversity and retrogressed stock resource within this species. (4) It is still unclear about geographic origination, molecular characterization and anthropogenic disturbance of various P. yezoensis cultivars as genetic resources from the viewpoint of global commercial utilization. Several molecular studies have identified the cultivated and wild strains of P. yezoensis through detecting genetic divergence (Kunimoto et al. 1999, 2003; Mizukami et al. 1999; Niwa and Aruga 2006; Niwa et al. 2009), but so far no clear clarification presented among all cultivated and wild strains of P. yezoensis throughout the world. In order to keep and advance the development of nori breeding, it is a pressing task to unambiguously and correctly elucidate the network relationships for most commonly used various P. yezoensis strains.
The internal transcribed spacer (ITS) of nuclear ribosomal DNA (nrDNA) is part of the rDNA cistron, which consists of 18S, ITS1, 5.8S, ITS2, and 26S. For over a decade, nrDNA ITS1 has been extensivey applied to reveal the phylogenetic relationships, discriminate wild and cultivated Porphyra blades (Mizukami et al. 1999; Niwa and Aruga 2003; Hu et al. 2007; Neefus et al. 2008; Niwa et al. 2009), and determine the origin of different cultivated Porphyra strains as well (Kunimoto et al. 1999). Although ITSs are found in thousands of copies within plant genomes, intra-genome diversity is generally low (Baldwin et al. 1995), which attributed to concerted evolution of intra- and inter-chromosomal loci (Baldwin et al. 1995; Ainouche and Bayes (1997); Won and Renner 2005), a process that acts through gene conversion and unequal crossing over. In addition, accumulating evidence suggests that intra-individual variation of nrDNA ITS regions should not be considered as exceptional (Feliner et al. 2004), and as a result of concerted evolution the occurrence of ancestral polymorphisms is not the most likely ultimate cause for intra-genomic variability in this marker. Here, we endeavored to include cultivars with doubtless breeding backgrounds, including more recently introduced P. yezoensis specimens in the Northwestern America from Asia. The primary objectives of this study were to determine the selection network relationships of various P. yezoensis cultivars worldwide using nrDNA ITS1 sequences; and to provide genetic insights for further artificial breeding and selection practices of Porphyra.
Materials and methods
Fifty-nine well-defined and published nrDNA internal transcribed spacer sequences (ITS1, length ≥345 bp) retrieved from GenBank/EMBL (sequences deposited by January 2009). GenBank accession numbers can be found in Kunimoto et al. (1999), Niwa and Aruga (2003), Hwang et al. (2005), Hu et al. (2007), Niwa et al. (2008) and Neefus et al. (2008). Based on the responding reference information, 59 P. yezoensis samples were sorted as wild type (7 samples), introduced type (21 samples) and cultivated type (31 samples), respectively. Information on sequence ID, sort type, location and reference are shown in Table 1. The boundaries of the coding and spacer regions were determined according to previously published Porphyra sequences (Hu et al. 2007; Niwa et al. 2008). The lengths of the ITS1 sequences range from 345 bp in a wild strain (Seq. ID H8) to 354 bp in one wild strain (Seq. ID H9) and six generated genotypes (Seq. ID H3, H13–H16, H23) of P. yezoensis (Table 1). Since the correction of PCR artifacts for the sequence of cloned PCR fragment is an important task in the search for patterns and extent of molecular evolution, in this study these issues can be addressed by clustering the sequences into 99% sequence similarity groups as a standard (Acinas et al. 2005).
Multiple alignments of the 59 representative sequences was performed via the program Clustal × 1.83 (Thompson et al. 1997) and then refined by eye. Identical sequences were represented by a single sequence in the alignments. Genotypes were assembled into separate groups according to origin or sort type. Nucleotide polymorphism as measured by θ w (Watterson 1975) and diversity as measured by π (Nei 1978), were calculated with DnaSP 4.5 (Rozas and Rozas 1999). Phylogenetic relationships of the 59 entities were conducted using neighbor joining (NJ), maximum parsimony (MP) and maximum likelihood (ML) methods as implemented in PAUP 4.0b10 (Swofford 2002). Specifically, MP analysis was performed using the heuristic search option, 50 random sequence additions, and the tree bisection reconnection (TBR) branch swapping. All sites were treated as equally weighted. For ML analysis, the model of nucleotide evolution that best fitted the data was determined by the hierarchical likelihood ratio test approach (hLRTs; Huelsenbeck and Crandall 1997) using Modeltest 3.7 (Posada and Crandall 1998). The defined model was the K80 model with the gamma distribution (shape parameter = 0, indicated equal rates for all site). ML analysis was carried out using the heuristic search option with random sequence addition (1,000 replicates). Bootstrap support values (Felsenstein 1985) were also estimated from 1,000 replications with 100 random addition replicates and TBR branch swapping algorithm for MP and ML analyses. Since the genealogical network analyses are powerful methods for intra-specific data in revealing multiple connections between genotypes and theoretically indicating the missing mutational connections (Posada and Crandall 2001), but traditional phylogenetic methods did not perform properly due to few substitutions. A genotype network with the option treating gaps as a fifth base was constructed based on the 95% parsimony criteria using the TCS 1.21 (Clement et al. 2000) to illustrate the mutational connections and the frequency of different genotypes among the various P. yezoensis strains worldwide.
Results
Partial nrDNA ITS1 region (345–354 bp) analyses were conducted on 360 aligned sites of 59 P. yezoensis samples. Sequence comparisons of the ITS1 fragment revealed 10 singleton variable sites and 42 polymorphic sites (15 parsimony-informative) with 15 transitions, 10 transversions and 17 indels. When sequence length was taken into consideration, the ITS1 region defined 23 genotypes (H1–H23), and of which 18 (78.26%) are unique in species P. yezoensis (Tables 1, 2). For the remaining five genotypes, two most common (H1 and H3) are present in 28.81 and 23.73% of the 59 specimens, respectively. Interestingly, among the 21 introduced samples in USA, 15 samples from New York, Massachusetts, Maine and New Hampshire generated one single genotype (H1) which has identical ITS1 sequence to two samples of the wild P. yezoensis strain (Ogatsu and NA-2) (Table 1). In addition, three samples from Texas, USA produced an endemic genotype H10. The ITS1 genotype H3 from Massachusetts, USA was identical to that of 13 cultivated strains from Japan (Table 1). Besides the above mentioned genotypes, we found different genotypes of P. yezoensis existed in one location: four genotypes generated from wild Ogatsu strains (OG-1, OG-2, OG-4 and Ogatsu), and three genotypes originated from wild Nanaehama strains (Nanaehama, NA-2 and NA-4), whereas five unique genotypes generated from cultivated P. yezoensis strains from Jiangsu, China (H3 and H13–H16) (Table 1), and this kind of genetic variability previously had also been detected by Kunimoto et al. (1999). When gaps treated as pairwise deletion, the pairwise distances among P. yezoensis genotypes ranged from 0 to 4.5% (0–15 bp), with zero percent indicating genotypes identified only by length.
The overall estimate of nucleotide polymorphism is presented in Table 3. In view of sort type, nearly 7-fold level of polymorphism was observed, with the highest in cultivated strains (π = 1.664 × 10−2, θ w = 2.011 × 10−2) and the lowest in wild strains (π = 0.253 × 10−2, θ w = 0.255 × 10−2). Again with a view to origin, South Korea exhibited the highest nucleotide variability (π = 1.437 × 10−2, θ w = 1.655 × 10−2) whereas Japan displayed the lowest diversity (π = 0.572 × 10−2, θ w = 0.597 × 10−2) in which is distributed with the maximum genotypes.
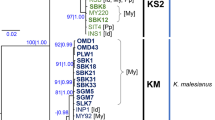
Based on the 23 nrDNA ITS1 sequences of P. yezoensis genotypes, a phylogenetic tree generated by NJ, MP and ML methods, recognized two clades with high bootstrap values (NJ = 99%, MP = 99%, ML = 95%) (Fig. 1). One clade is comprised of 15 P. yezoensis genotypes, of which five (wild type) from Japan and eight (cultivated type) from China and Japan; the other clade consists of five genotypes (cultivated type) from South Korea and China, and three genotypes (introduced type) of which one is endemic to Texas, USA and the rest two are cultured in University of Connecticut, USA, but originally introduced from Qingdao, China. The genotype variation “treating gaps as the fifth character” with genealogy parsimony analysis also showed two clades in P. yezoensis strains with similar topological structure (Fig. 2). Table 2 shows the character states at 22 and 10 nucleotide positions of the nrDNA ITS1 region for 13 and 10 genotypes in P. yezoensis, respectively. Overall, single mutation (including indel) was observed at 11 sites (31.43%) and was not shared among specimens. Likewise, four character indels (sites 92–93, 242–244, 347 and 349) were found in all genotypes after alignment (Table 2), which can discriminate the 23 genotypes into two separate clades.

Neighbor-joining tree of 23 Porphyra yezoensis genotypes generated from 360 sites of nrDNA ITS1 data set. Numbers given on each branch represent bootstrap values (>50%) for NJ (left), MP (middle) and ML (right) analyses

TCS statistical parsimony network of 23 ITS1 genotypes of Porphyra yezoensis with treating gaps as the fifth character. Lines connecting the genotypes represent a single mutation with solid circle representing inferred mutational steps not observed in this study. Diameters of the circles are proportional to genotype size
Discussion
So far, intra-specific selective breeding is the dominant approach for nori breeders to obtain strains with excellent production traits. In the present study, molecular data revealed some cryptic intra-specific genetic diversity in P. yezoensis strains. Although we examined only 2–5 specimens in many areas, nrDNA ITS1 strongly favored wild P. yezoensis strains with higher genetic diversity in Japan and demonstrated other primary diversity centers of cultivated strains in China and South Korea (as well as higher nucleotide polymorphism, π = 0.734 × 10−2 and 1.437 × 10−2, respectively), which are useful genetic insights for future commercial breeding of P. yezoensis on a global scale. The nrDNA ITS1 revealed that four distinct P. yezoensis genotypes corresponded to four wild strains from Miyagi Prefecture, Japan, and also three unique genotypes corresponding to three wild specimens from Hokkaido, Japan (Table 1). These showed significantly higher intra-specific genetic diversity in wild species P. yezoensis in the two areas despite small nucleotide variation (π = 0.253 × 10−2, θ w = 0.255 × 10−2). Therefore genetically, the seven wild strains from both Miyagi and Hokkaido areas can be used target parent breeding materials for selective or cross-breeding in the future. Significantly, these seven wild P. yezoensis strains should be exceptionally preserved as crucial stock resource in order to keep sustainable development and utilization of economic seaweeds. Besides, phylogenetic analyses indicated 23 various P. yezoensis genotypes clustered clearly as two clades (Figs. 1, 2), in which five genotypes (H1, H6, H7, H8 and H9, they were generated from six wild strains) exhibited closest relationship to one genotype (H4, it was originated from a cultivated strain in Japan). Accordingly, these six wild P. yezoensis strains can potentially be utilized for cross-breeding with other cultivated strains in Japan, China and South Korea, and some introduced areas in USA as well for mariculture practices. Remarkably, cultivated P. yezoensis strains from both China and South Korea displayed higher genetic diversity with some distinct genotypes in each area, together with higher nucleotide variability (South Korea: π = 1.437 × 10−2, θ w = 1.655 × 10−2; China: π = 0.734 × 10−2, θ w = 0.678 × 10−2) (Table 3), and hence selective breeding and cross-breeding can be carried out each other among them through exchange of demanding cultivars, certainly exchanging with wild P. yezoensis strains from Japan as well. But it should be more cautious for intensive cross-breeding among the seven cultivated strains (corresponding genotypes H11, H12, H17–H21), because both phylogenetic relatedness and parsimony genealogy showed the closest relationships among them (Figs. 1, 2). In contrast, considerably high genetic uniformity existed in various cultivated strains in Hyogo Prefecture, Japan (Table 1). Similarly, the ITS1 sequence demonstrated that the five cultivated P. yezoensis strains (Ariake-1, D-18-1, Obagreen, Sasiki and Saga-5) in Japan are actually the same stock resource. As a consequence, cross-breeding should be furthest refrained among these cultivated strains to prevent declined genetic diversity.
Unsurprisingly, three phylogenetic approaches detected that two introduced P. yezoensis genotypes (H11, H12) currently cultured in University of Connecticut were closely assembled with one cultivated genotype (H21) (Fig. 1). Exceptionally, the introduced cultivars of P. yezoensis in USA contained five specific genotypes and exhibited higher nucleotide diversity (π = 1.043 × 10−2, θ w = 0.974 × 10−2), which guarantees the genetic feasibility of extensive breeding and farming in USA for this economic species. In addition, DNA data has been proved useful to pursue the introduced origin of P. yezoensis in Northwest Atlantic (Neefus et al. 2008). In this study, the three genotypes (H11–H12, H21) grouped closely (Fig. 1), together with a few exceptional nucleotide substitutions like sites 68, 128, 192 and 279 (Table 2). So we believe that the ITS1 sequences patterns have the potential to track the geographic origin of various specimens of P. yezoensis, which is consistent with one previous report (Kunimoto et al. 1999). If this suppose is correct, the introduced genotype (H10) from two locations in Texas, USA also clustered with the three genotypes, which probably ascribed to an unintentional anthropogenic introduction from Qingdao, China through maritime traffic.
Inter-specific cross breeding is another widespread method to produce strains meeting the breeder’s demand, and some examples have been documented in brown macroalgae (Coyer et al. 2002, 2007; Wallace et al. 2004), but to date no reports have been published on natural inter-specific hybridization in Porphyra. Although P. yezoensis is a more boreal species than P. tenera Kjellman (Miura and Aruga 1987; Miura 1988), previous investigations indicated that the inter-specific hybridization between the two species might ever occurred in Northeastern Japan because they have a partial overlapped distribution zone in this area (Miura and Aruga 1987; Miura 1998; Yoshida 1998, 2000). The latest molecular study about Porphyra species demonstrated that plastid introgression occurred from P. yezoensis to P. tenera (Niwa et al. 2009), and one specimen (MT-1) was even proposed as a progeny of inter-specific hybridization between the two species (Niwa et al. 2009). Hence, inter-specific hybridization of Porphyra will become an important direction of developing new strains for the future.
Currently indecisive parentage pedigree largely embarrassed the breeding progress of P. yezoensis. Over twenty cultivated P. yezoensis strains included in this study did not have specific genetic background information. These strains were distributed in different areas in East Asia revealing their genetic relationships with other wild and cultivated strains. For example, the Korean samples (genotypes H17–H20) and the Chinese samples (H11, H12 and H21) are closely grouped together (Figs. 1, 2), which suggested these currently farmed P. yezoensis strains in Korea and China not only were introduced from Japan (Hwang et al. 2005), but also were the same parentage pedigree. Likewise, generally old genotypes with wide distribution are located internally in the network tree (Posada and Crandall 2001; Keirstein et al. 2004), as with genotype H1 and H3 in this study (Fig. 2) which might be the most common used parentage materials for selective breeding or P. yezoensis. This clustering analysis based on the nrDNA ITS1 region can resolve the genetic background issues of the cultivars with unknown pedigree. Summarily, the relationships of P. yezoensis strains with known parentage will help nori breeders to select appropriate parents in their breeding practices to maximize yield as well as to maintain genetic diversity. In the end, we admit that as a single gene region, nrDNA ITS1 can not explicitly discover the intrinsic genealogical relationships among strains within P. yezoensis, and integrated locus analyses like mtDNA Cox1 is desirable to obtain reliable genetic inference in future.
References
Acinas SG, Sarma-Rupavtarm R, Klepac-Ceraj V, Polz MF (2005) PCR-induced sequence artifacts and bias: insights from comparison of two 16S rRNA clone libraries constructed the same sample. Appl Environ Microbiol 71:8966–8969
Ainouche ML, Bayes RJ (1997) On the origins of the tetraploid Bromus species (section Bromus, Poaceae): insights from the internal transcribed spacer sequences of nuclear ribosomal DNA. Genome 40:730–743
Baldwin BG, Sanderson MJ, Porter JM, Wojciechowski MF, Campbell CS, Donoghue MJ (1995) The ITS region of nuclear ribosomal DNA: a valuable source of evidence on angiosperm phylogeny. Ann Mo Bot Gard 82:247–277
Clement M, Posada D, Crandall K (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1659
Coyer JA, Peters AF, Hoarau G, Stam WT, Olesn JL (2002) Hybridization of the marine seaweeds, Fucus serratus and F. evanescens (Heterokontophyta: Phaeophyceae) in a century-old zone of secondary contact. Proc R Soc Lond Biol Sci 269:1829–1834
Coyer JA, Hoarau G, Stam WT, Olsen JL (2007) Hybridization and introgression in a mixed population of the intertidal seaweeds Fucus evanescens and F. serratus. J Evol Biol 20:2322–2333
Feliner GN, Larena BG, Aguilar JF (2004) Fine scale geographic structure, intra-individual polymorphism and recombination in nuclear ribosomal internal transcribed spacers in Armeria (Plumbaginaceae). Ann Bot 93:189–200
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–793
Hu ZM, He YJ, Xia P, Duan DL (2007) Molecular identification of Chinese cultivated Porphyra (Bangiaceae, Rhodophyta) based on the rDNA internal transcribed spacer-1 sequence and random amplified polymorphic DNA markers. Mar Biol Res 3:20–28
Huelsenbeck JP, Crandall KA (1997) Phylogeny estimation and hypothesis testing using maximum likelihood. Ann Rev Ecol Syst 28:437–466
Hwang MS, Kim SM, Ha DS, Baek JM, Kim HS, Choi HG (2005) DNA sequences and identification of Porphyra cultivated by natural seeding on the Southwest coast of Korea. Algae 20:183–196 (in Korean with English abstract)
Iitsuka O, Nakamura K, Ozaki A, Okamoto N, Saga N (2002) Genetic information of three pure lines of Porphyra yezoensis (Bangiales, Rhodophyta) obtained by AFLP analysis. Fish Sci 68:1113–1117
Keirstein G, Vallinoto M, Silva A, Schneider MP, Iannuzzi L, Brenig B (2004) Analysis of mitochondrial D-loop region casts new light on domestic water buffalo (Bubalus bubalis) phylogeny. Mol Phylogenet Evol 30:308–324
Kunimoto M, Kito H, Kaminishi Y, Mizukami Y, Murase N (1999) Molecular divergence of the SSU rRNA gene and internal transcribed spacer 1 in Porphyra yezoensis (Rhodophyta). J Appl Phycol 11:211–216
Kunimoto M, Kito H, Mizukami Y, Murase N, Levine I (2003) Molecular features of a defined genetic marker for the determination of the Porphyra tenera lineage. J Appl Phycol 15:337–343
Miura A (1988) Taxonomic studies of Porphyra species cultivated in Japan, referring to their transition to the cultivated variety. J Tokyo Univ Fish 75:311–325
Miura A (1998) Asakusa-nori: data book of rare aquatic animals and plants of Japan, edited by Japan Fisheries Resource Conservation Association. Japan Fisheries Resource Conservation Association, Tokyo, pp 298–299 (in Japanese)
Miura A, Aruga Y (1987) Distribution of Porphyra in Japan as affected by cultivation. J Tokyo Univ Fish 74:41–50
Mizukami Y, Kito H, Kaminishi Y, Murase N, Kunimoto M (1999) Nucleotide sequence variation in the ribosomal internal transcribed spacer regions of cultivated (cultivars) and field-collected thalli of Porphyra yezoensis. Fish Sci 65:788–789
Neefus CD, Mathieson A, Bray TL, Yarish C (2008) The distribution, morphology, and ecology of three introduced Asiatic species of Porphyra (Bangiales, Rhodophyta) in the Northwestern Atlantic. J Phycol 44:1399–1414
Nei M (1978) Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89:583–590
Nei M, Li WH (1979) Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc Natl Acad Sci USA 76:5269–5273
Niwa K, Aruga Y (2003) Rapid DNA extraction from conchocelis and ITS-1 rDNA sequences of seven strains of cultivated Porphyra yezoensis (Bangiales, Rhodophyta). J Appl Phycol 15:29–33
Niwa K, Aruga Y (2006) Identification of currently cultivated Porphyra species by PCR-RFLP analysis. Fish Sci 72:143–148
Niwa K, Kikuchi N, Iwabuchi M, Aruga Y (2004) Morphological and AFLP variation of Porphyra yezoensis Ueda form narawaensis Miura (Bangiales, Rhodophyta). Phycol Res 52:180–190
Niwa K, Kato A, Kobiyama A, Kawai H, Aruga Y (2008) Comparative study of wild and cultivated Porphyra yezoensis (Bangiales, Rhodophyta) based on molecular and morphological data. J Appl Phycol 20:261–270
Niwa K, Iida S, Kato A, Kawai H, Kikuchi N, Kobiyama A, Aruga Y (2009) Genetic diversity and introgression in two cultivated species (Porphyra yezoensis and Porphyra tenera) and closely related wild species of Porphyra (Bangiales, Rhodophyta). J Phycol 45:493–502
Posada D, Crandall KA (1998) Modeltest: testing the model of DNA substitution. Bioinformatics 14:817–818
Posada D, Crandall KA (2001) Intraspecific gene genealogies: trees grafting into networks. Trends Ecol Evol 16:37–45
Rozas J, Rozas R (1999) DnaSP version 3: an integrated program for molecular population genetics and molecular evolution analysis. Bioinformatics 15:174–175
Swofford DL (2002) PAUP: phylogenetic analysis using parsimony (and other methods). Version 4.0b10. Sinauer, Sunderland
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The Clustal X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 24:4876–4882
Wallace A, Klein AS, Mathieson AC (2004) Determing the affinities of salt march fucoids using microsatellite markers: evidence of hybridization and introgression between two species of Fucus (Phaeophyta) in a Maine estuary. J Phycol 40:1013–1027
Watterson GA (1975) On the number of segregating sites in genetical models without recombination. Theor Pop Biol 7:256–276
Weng ML, Liu B, Jin DM, Yang QK, Zhao G, Ma JH, Xu P, Duan DL, Wang B (2005) Identification of 27 Porphyra lines (Rhodophyta) by DNA fingerprinting and molecular markers. J Appl Phycol 17:91–97
Won H, Renner SS (2005) The internal transcribed spacer of nuclear ribosomal DNA in the gymnosperm Gnetum. Mol Phylogenet Evol 36:581–597
Yoshida T (1998) Marine algae of Japan. Uchida Rokakuho, Tokyo, p 222 (in Japanese)
Yoshida T (2000) Porphyra tenera Kjellman: threatened wildlife of Japan, edited by Environment Agency of Japan, 2nd edn. Japan Wildlife Research Center, Tokyo, p 218 (in Japanese)
Acknowledgments
The authors thank anonymous reviewers for insightful criticisms and comments on an earlier version of this manuscript. This research was partly supported by China Postdoctoral Science Foundation project (No. 20090451356) and Knowledge Innovation Program of the Chinese Academy of Sciences on Biodiversity Project.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hu, Z., Liu, F., Shao, Z. et al. NrDNA internal transcribed spacer revealed molecular diversity in strains of red seaweed Porphyra yezoensis and genetic insights for commercial breeding. Genet Resour Crop Evol 57, 791–799 (2010). https://doi.org/10.1007/s10722-009-9519-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10722-009-9519-y