
Abstract
This study focused on clarifying phylogenetic relationships among Citrus accessions from Vietnam. Our phylogenetic analysis based on nucleotide sequences from the ITS of the ribosomal DNA included 69 accessions belonging to Citrus and related (sub)genera. Maximum parsimony and Bayesian analysis confirmed a clear separation of the three ‘true’ Citrus species (C. medica, C. maxima and C. reticulata). Confirming recent taxonomic revisions, Fortunella, Poncirus trifoliata and Citrus hystrix are clustered among the accessions of subgenus Citrus. C. × sinensis accessions revealed a close evolutionary relationship to either C. maxima or C. reticulata, thereby confirming their involvement in its hybrid origin. Also, some other hybrid taxa and their proposed parental species were investigated and their origin could in some cases be confirmed using the ITS sequence data.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The genus Citrus is one of 33 (Swingle and Reece 1967) to 26 (Mabberley 2008a, b) genera in the subfamily Aurantioideae of the family Rutaceae. Citrus is divided into two subgenera: the common cultivated types of this fruit are placed in the subgenus Citrus, species of the subgenus Papeda do not bear edible fruit (Moore 2001).
The taxonomy of Citrus is complex and the precise number of natural species is unclear. Until the mid 1970s, Citrus taxonomy was based solely on morphological and geographical data, leading to two widely used classification systems. The Swingle system (Swingle and Reece 1967) is relatively simple, containing 16 species, but the Tanaka taxonomy recognizes up to 162 species (Tanaka 1977). This lack of agreement reflects differences of opinion as to what degree of difference justifies species status and Tanaka has split the genus Citrus into many small groups. There is no definitive work on Citrus taxonomy, and many scientists use a system intermediate between these two systems (Hodgson 1965, 1967: 36 species; Singh and Nath 1969: 31 species).
Scora (1975) and Barrett and Rhodes (1976) suggested that there are only three (or four) ‘basic’ true species within the subgenus Citrus (as defined by Swingle): citron (C. medica), mandarin (C. reticulata), and pummelo (C. maxima), and sometimes lime (now generally considered a hybrid, C. × aurantiifolia). Other cultivated Citrus taxa within the subgenus Citrus are believed to be hybrids derived from these true species, species of the subgenus Papeda, or closely related “genera”. Interestingly, the earliest plant taxonomists also believed that there were only few valid species of the subgenus Citrus (two species, and three varieties in Linnaeus 1753; three species Hooker 1875) and this idea has gained scientific support in recent years from molecular data (Federici et al. 1998; Nicolosi et al. 2000). For instance, morphological and molecular studies have indicated that lime (C. × aurantiifolia) and lemon (C. × limon) arose from interspecific crosses with C. medica (citron) as one of the parent species (Scora 1975; Barrett and Rhodes 1976; Federici et al. 1998; Nicolosi et al. 2000).
Genetic variability and relationships among cultivated taxa is complicated by several factors, like the high frequency of bud mutation and nucellar embryony, a long history of cultivation and wide cross-compatibility, leading to taxonomic ambiguities (Nicolosi et al. 2000; Moore 2001). Spontaneous or artificial hybridization and sport formation has probably played an important role in the origin of many cultivated Citrus taxa. The wide human-induced dispersion of Citrus across geographic boundaries has further facilitated “intergeneric” and intrageneric crossing leading to an immense variety of morphological forms. Interesting hybrids or sports can easily be vegetatively propagated, leading to a contrast between high levels of morphological (mainly agronomic) trait diversity versus low levels of genetic variability within taxa, as described for clementine (C. reticulata Blanco; Bretó et al. 2001); lemon (C. × limon (L.) Osbeck.; Gulsen and Roose 2001) and trifoliate oranges (Poncirus trifoliata; Fang et al. 1997).
Understanding taxonomy, phylogenetic relationships and genetic variability in Citrus is critical for determining genetic relationships, characterizing germplasm, establishing breeding programs and the registration of new cultivars. Vietnam, located in the South East Asian center of origin of Citrus (Webber 1967; Scora 1975), is a center of biodiversity for wild and cultivated Citrus accessions (Tanaka 1954). Citrus has always been one of Vietnam’s most popular fruit products and as a consequence they are grown widely from the North to the South of Vietnam, leading to a high abundance of Citrus genetic resources.
In this study the phylogenetic relationships among 61 Citrus accessions collected in Vietnam were analysed using ITS sequences of the rDNA, and compared with data from 8 accessions present in the NCBI-database.
Materials and methods
Plant material
Table 1 gives an overview of the analysed accessions, their origin and institute codes. Table 2 provides the scientific names of all taxa used in this manuscript, corresponding vernacular names and their names as put forward by Mabberley (2008a, b).
In total 51 accessions belonging to Citrus subgenus Citrus were collected: 1 C. × paradisi (grapefruit: G), 11 C. maxima (pummelo: P), 5 C. medica (citron: C), 3 C. × aurantiifolia (lime: L), 1 C. × limon (lemon: LI), 12 C. × sinensis (sweet orange: O), 1 C. × aurantium (sour orange: OS), 15 C. reticulata (mandarin: M), 1 C. reticulata ‘Clementine’ (Clementine: M), 1 C. × nobilis (king mandarin: MK). Furthermore, the following 10 accessions, that have been described to belong to closely related genera or subgenera, were included in the sample set: 5 specimens of C. hystrix (kaffir lime: KL), belonging to the subgenus Papeda, 1 Poncirus trifoliata accession (PT), 3 Fortunella accessions (Fortunella japonica and Fortunella margarita: F) and 1 Murraya paniculata specimen.
Next to the 61 specimens collected in Vietnam, the NCBI database contains ITS sequence data from 6 Citrus accessions, and from 2 accessions from the related genus Murraya: Murraya paniculata and Murraya koenigii.
DNA isolation
Total cellular DNA was isolated as described by Rogers and Bendich (1988) with minor modifications. Young leaves from fully expanded and mature plants were collected and maintained at low temperature in polyethylene bags. In the laboratory, the leaves were washed in distilled water, ethanol 70% and ground using the Retsch mixer mill model MM 200. Each sample was suspended in 1.0 ml of DNA extraction buffer. After incubation at 65°C for 30 min with occasional vigorous shaking, the samples were centrifuged at 13,000g for 10 min. The supernatant was collected and an equal volume (about 700 μl) of iso-propanol was added. The samples were mixed, and placed on ice (or −20°C) for 2 h. The samples were centrifuged at 13,000g for 10 min and the supernatant was discarded. After addition of 400 μl of TE Buffer and 5 μl RNase the samples were incubated at 37°C for 20 min. 400 μl of CTAB Buffer was added and the samples were transferred to a warm water bath at 65°C for 15 min. Afterwards, an equal volume of isoamyl alcohol:chloroform (24:1) was added, and the samples were centrifuged at 13,000g. To the aqueous phase (upper phase) two volumes of 96% ethanol were added. After incubation at room temperature for 5 min the samples were centrifuged for 5 min (10,000g). The pellet was then washed twice with ethanol 70%. The DNA was resuspended in 200 μl TE Buffer, applying a short incubation at 37°C. DNA samples were stored at −20°C.
DNA extraction, PCR and sequencing
PCR amplification of the ITS region, including the 5.8 S rDNA region, was performed using primers ITS-1 and ITS-4 (ITS1: 5′ TCCGTAGGTGAACCTGCGG 3′; ITS4: 5′ TCCTCCGCTTATTGATATGC 3′ as described by White et al. (1990), using a Perkin Elmer 9700 thermal cycler (Applied Biosystems corporation). Final reaction volumes of 25 μl each contained 50 ng genomic DNA, 0.5 μM of each primer, 0.2 mM dNTPs, 0.5 U Taq DNA polymerase (Fermentas), 1× PCR buffer supplied by the manufacturer and about 2.5 mM MgCl2. The amplification programme consisted of predenaturation at 94°C for 90 s; 30 cycles at 95°C for 50 s, 55°C for 70 s and 72°C for 90 s; and a final incubation at 72° for 3 min; 1 min at 30°C; and a final hold at 4°C. MgCl2 concentration and annealing temperature had to be optimized for some of the samples to obtain a good amplification.
PCR products were purified by PureLinkTM PCR Purification kit (Invitrogen). Purified fragments were directly sequenced with PCR primers using the ABI prism BigDyeTM Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) on an automated sequencer (ABI prism 3130, Applied Biosystems).
Phylogenetic analyses
Next to the 61 new sequences obtained from Vietnamese accessions in the current analysis, ITS sequence data from 6 Citrus accessions, and from 2 accessions from related genus Murraya (Murraya paniculata and Murraya koenigii) was included in the dataset.
Sequences were aligned using ClustalX (Thompson et al. 1997) followed by manual adjustments using BioEdit 7.0.5.3. Phylogenetic analyses were carried out using PAUP* v4.0b10 (Swofford 2002) and MrBayes 3.2.1 (Ronquist and Huelsenbeck 2003). Parsimony analyses were performed with PAUP* v4.0 b10 (Swofford 2002) using the heuristic search option with random sequence addition (100 random replications) and TBR branch-swapping. All characters had equal weight, and gaps were treated as missing characters. Constant and uninformative characters were removed from the data matrix. Consistency index (CI), retention index (RI) and rescaled consistency index (RC) were calculated. Support for the different clades was tested by bootstrap analysis (100 replicates using heuristic search, simple sequence addition). Bayesian analysis was run using MrBayes version 3.1.2. (Ronquist and Huelsenbeck 2003). Bayesian inference was run for 3,000,000 generations, and the first 100,000 generations were discarded as burn-in.
Results
Sequence data from the ITS region of the rDNA was analysed for 69 accessions, including 8 sequences obtained from the NCBI database. The alignment of all sequences included 703 positions (including gaps), 298 positions were variable among which 126 were parsimony-informative sites.
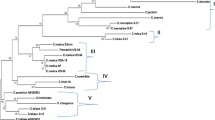
Parsimony analysis produced 14,778 equally parsimonious trees of 286 steps with a consistency index (CI) of 0.6538, a retention index (RI) of 0.8350, and a rescaled consistency index (RC) of 0.5460. A majority-rule consensus tree was constructed from these trees as shown in Fig. 1. The bootstrap values (100 replicates) are shown on each branch. The tree reconstructed by Bayesian analysis is shown in Fig. 2. The tree obtained is very similar to that of the parsimony method, although the separation of the subclusters is less obvious in the Bayesian tree.

Majority rule consensus tree of 14,778 maximum parsimonious trees of 69 Citrus accessions based on ITS of the rDNA sequence data. Tree length = 286; consistency index (CI) = 0.6538; retention index (RI) = 0.8350; rescaled consistency index (RC) = 0.5460. Bootstrap values above 40% are given on the nodes. The tree is rooted with the three Murraya accesssions

Bayesian phylogenetic tree of 69 Citrus accessions based on ITS of the rDNA sequence data. The posterior probability is given on each node. The tree is rooted with the three Murraya accesssions. The scale bar represents branch length (number of substitutions/site)
The phylogenetic trees based on both maximum parsimony and Bayesian analyses show a clear separation between the three ‘basic’ species as proposed by Scora (1975) and Barrett and Rhodes (1976). Cluster 1 contains all C. maxima accessions together with C. × paradisi, 1 C. × aurantiifolia (1a), and Poncirus trifoliata and some of the C. × sinensis genotypes (1b). Cluster 2 combines C. medica with all other C. × aurantiifolia and C. × limon (2a); and C. hystrix (2b). C. reticulata and C. × aurantium are grouped (3a), together with most of the C. × sinensis. Fortunella japonica and Fortunella margarita are in 3b. Pairwise sequence divergence ranged from 0 (between multiple accessions) to 0.215 (between Murraya koenigii and Cmedica1) with an average of 0.038. Within Citrus, average pairwise sequence divergence was 0.030, with a maximum of 0.143 (Cmedica1 vs. 043Xadoai). No sequence divergence was found among a few mandarin accessions from cluster 3a, and among some oranges from the same cluster. 3 C. medica accessions (cluster 2a) also revealed 100% ITS sequence identity. Furthermore, 7 C. maxima members of cluster 1 have identical ITS sequences. Interestingly, ITS sequence of the grapefruit accession was 100% identical to these C. maxima accessions.
Discussion
In this study sequence data from ITS of the rDNA of 69 accessions from the genus Citrus and related genera were obtained and their evolution was investigated using maximum parsimony and Bayesian analyses. In contrast to previous studies the current phylogenetic analysis includes a larger number of closely related accessions of a few closely related species instead of one (or a few) specimens from different genera, which allows us to investigate relationships at a lower taxonomic level and to investigate evolutionary divergence within taxa.
The separation of the three ‘true’ Citrus (C. medica, C. maxima and C. reticulata) is confirmed by their grouping in three different clusters in our ITS of the rDNA sequence analysis.
As isozyme and morphological data suggested before (Barrett and Rhodes 1976; Torres et al. 1978) and could be expected based on vegetative propagation of cultivars, ITS-data reveal a close evolutionary relationship among the analysed C. maxima accessions. Furthermore, our data confirm a close evolutionary relationship between C. maxima, C. × paradisi and, although more distantly, some sweet orange accessions (C. × sinensis). cpDNA analysis of these three species has also shown these species to be very closely related (Nicolosi et al. 2000; Kyndt et al., unpublished data).
Grapefruit (C. × paradisi) has been proposed to be of hybrid origin, with pummelo as mother and sweet orange as father (Gmitter 1995; Moore 2001) and subsequent backcrossing with pummelo (Fang and Roose 1997; Herrero et al. 1996; Pang et al. 2007, Mabberley 2008a, b). This hypothesis is confirmed by our ITS sequence data, since the grapefruit accession reveals 100% sequence identity with some pummelo accessions at ITS level.
The mandarin (C. reticulata) cluster is not well resolved, as also seen in other molecular data analyses (Federici et al. 1998; Barkley et al. 2006). Sour oranges (C. × aurantium) and most of the sweet oranges (C. × sinensis) cluster among the mandarins, confirming the mandarins as one of their parental species (Barrett and Rhodes 1976) as suggested by previous molecular data (Nicolosi et al. 2000; Barkley et al. 2006; Pang et al. 2007).
Sweet orange (C. × sinensis) is thought to be a natural hybrid between predominantly C. reticulata and some C. maxima traits (Scora 1975; Barrett and Rhodes 1976). Molecular data already confirmed that the chloroplast genome of sweet orange is derived from pummelo (Green et al. 1986; Nicolosi et al. 2000; Barkley et al. 2006; Kyndt et al., unpublished data). This ITS-sequence analysis and the chloroplast PCR-RFLP study of Jena et al. (2009) suggest that C. × sinensis has a polyphyletic origin. While some Vietnamese sweet orange genotypes are closely related with pummelo, others are grouped with mandarin. Most probably C. × sinensis originated from one or a few hybridization events between pummelo as maternal parent and mandarin as father and subsequent backcrosses with one of these parents. It has to be noted that some well-established C. × sinensis cultivars (‘Xadoai’ and ‘Valencia’) are highly supported in the phylogenetic trees, and are found within the mandarin group.
Citrus medica is grouped with the proposed hybrid species C. × limon (lemon) and C. × aurantiifolia (lime), which is consistent with the fact that the citron has been proposed to be the paternal ancestor of several hybrids in Citrus (Federici et al. 1998; Nicolosi et al. 2000). While Barrett and Rhodes (1976) proposed that lime arose from a trihybrid “intergeneric” cross involving C. medica, C. maxima and a “Microcitrus” species, RAPD and SCAR markers (Nicolosi et al. 2000) suggested that limes resulted from a cross between C. micrantha (subgenus Papeda) and C. medica (male parent). Isozyme analyses (Torres et al. 1978) found low diversity between seven cultivars of lime (C. × aurantiifolia) and this was suggested to be due to its apomictic perpetuation (Barrett and Rhodes 1976). However, this is contradicted by the current study, where one lime accession clusters with pummelo and all others with citron, suggesting C. maxima and C. medica as parent species of C. × aurantiifolia.
Lemons (C. × limon) are thought to be natural hybrids of C. medica and lime (Scora 1975; Barrett and Rhodes 1976) or a hybrid of citron and sour orange (Gulsen and Roose 2001). Lemon is clustered among the citron-lime group (cluster 2a in Fig. 1), suggesting that lime is indeed one of the ancestors of lemon (Scora 1975; Barrett and Rhodes 1976). No relationship with sour orange is seen in our study. Although our ITS data confirm a close relationship between C. × limon and C. medica, and thereby confirm the involvement of the latter species in its hybrid origin, no clear-cut conclusions can be drawn about the other hypothetic ancestor species of this hybrid taxon.
ITS data shows a close evolutionary relationship between Fortunella and Citrus spp., although their morphology is very different. This observation agrees with previous molecular studies (Green et al. 1986; Pang et al. 2007), where some analyses even showed a nested clustering of Fortunella in Citrus (Herrero et al. 1996; Federici et al. 1998; Nicolosi et al. 2000; Pang et al. 2003; Barkley et al. 2006). Also in our analysis Fortunella spp. are clustered within Citrus, close to the C. reticulata group, confirming their recent reclassification as Citrus japonica (Mabberley 2008a, b). The same is true for Poncirus trifoliata, which is clustered within Citrus, and is now called Citrus trifoliata (Mabberley 2008a, b).
While Herrero et al. (1996) and Federici et al. (1998) find C. hystrix (kaffir lime, subgenus Papeda) clustered with C. maxima, Nicolosi et al. (2000) and our ITS data suggest that C. hystrix is closer to C. medica, C. × limon and C. × aurantiifolia. Generally, these observations demonstrate that the subdivision of the subgenera Citrus and Papeda, as proposed by Swingle and Reece (1967), based on the abundant presence of acridic oil in the fruit and the very broadly winged petioles in subgenus Papeda is not confirmed by molecular data. Based on all observations we can hypothesize that C. hystrix is a probable (grand)parent or sister species of C. × aurantiifolia or C. maxima and subsequently diverged independently from the subgenus Citrus.
References
Barkley NA, Roose ML, Krueger RR, Federici CT (2006) Assessing genetic diversity and population structure in a Citrus germplasm collection utilizing simple sequence repeat markers (SSRs). Theor Appl Genet 112:1519–1531
Barrett HC, Rhodes AM (1976) A numerical taxonomic study of the affinity relationships in cultivated Citrus and its close relatives. Syst Bot 1:105–136
Bretó MP, Ruiz C, Pina JA, Asins MJ (2001) The diversification of Citrus clementina Hort. Ex Tan., a vegetatively propagated crop species. Mol Phyl Evol 21:285–293
Fang DQ, Roose ML (1997) Identification of closely related Citrus cultivars with inter-simple sequence repeats markers. Theor Appl Genet 95:408–417
Fang DQ, Roose ML, Krueger RR, Federici CT (1997) Fingerprinting trifoliate orange germplasm accessions with isozymes, RFLPs, and inter-simple sequence repeat markers. Theor Appl Genet 95:211–219
Federici CT, Fang DQ, Scora RW, Roose ML (1998) Phylogenetic relationships within the genus Citrus (Rutaceae) and related genera as revealed by RFLP and RAPD analysis. Theor Appl Genet 94:812–822
Gmitter FG Jr (1995) Origin, evolution and breeding of the grapefruit. Plant Breed Rev 13:345–363
Green RM, Vardi A, Galune E (1986) The plastome of Citrus. Physical map, variation among Citrus cultivars and species and comparison with related genera. Theor Appl Genet 72:170–177
Gulsen O, Roose ML (2001) Lemons: diversity and relationships with selected Citrus genotypes as measured with nuclear genome markers. J Amer Soc Hort Sci 126:309–317
Herrero R, Asins MJ, Carbonell EA, Navarro L (1996) Genetic diversity in the orange subfamily Aurantioideae. I. Intraspecies and intragenus genetic variability. Theor Appl Genet 92:599–609
Hodgson RW (1965) Taxonomy and nomenclature in the Citrus fruits. In: Krishnamurthi S (ed) Advances in agriculture sciences and their applications. Agric. Coll. Res. Inst., Coimbatore, pp 317–331
Hodgson RW (1967) Horticultural varieties of Citrus. In: Reuther W, Webber HJ, Batchelor LD (eds) The Citrus industry. Vol. I. history world distribution, botany, and varieties. Univ of California, Div of Agricultural Sci, Berkeley, pp 431–591
Hooker JD (1875) Rutaceae. In: Reeve L (ed) The flora of British India. London, pp 484–517
Jena SN, Kumar S, Nair NK (2009) Molecular phylogeny in Indian Citrus L. (Rutaceae) inferred through PCR-RFLP and trnL-trnF sequence data of chloroplast DNA. Scientia Hort 119:403–416. doi:10.1016/j.scienta.2008.08.030
Linnaeus C (1753) Species plantarum, exhibentes plantas rite cognitas, ad genera relatas, cum differentiis specificis, nominibus trivialibus, synonymis selectis, locis natalibus, secundum systema sexuale digestas. Holmiae
Mabberley DJ (2008a) 21. CITRUS Linnaeus, Sp. Pl. 2: 782. 1753. Flora China 11:90–96
Mabberley DJ (2008b) Mabberley’s plant book: a portable dictionary of plants, 3rd edn. Cambridge University Press, Avon
Moore GF (2001) Orange and lemons: clues to the taxonomy of Citrus from molecular markers. Trends Genet 17:536–540
Nicolosi E, Deng ZN, Gentile A, La Malfa S, Continella G, Tribulato E (2000) Citrus phylogeny and genetic origin of important species as investigated by molecular markers. Theor Appl Genet 100:1155–1166
Pang XM, Hu DG, Deng XX (2003) Phylogenetic relationships among Citrus and its relatives as revealed by SSR markers. Acta Genet Sin 30:81–87
Pang XM, Hu CG, Deng XX (2007) Phylogenetic relationships within Citrus and its related genera as inferred from AFLP markers. Genet Resour Crop Evol 54:429–436
Rogers SO, Bendich AJB (1988) Extraction of DNA from plant tissues Plant molecular Biology Manual, vol A6. Kluwer Academic Publishers, Dordrecht, pp 1–10 printed in Belgium
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 12:1572–1574
Scora RW (1975) On the history and origin of Citrus. Bull Torrey Bot Club 102:369–375
Singh R, Nath N (1969) Practical approach to the classification of Citrus. Proceedings of 1st international Citrus symposium, pp 435–440
Swingle WT, Reece PC (1967) The botany of Citrus and its wild relatives. In: Reuther W, Webber HJ, Batchelor LD (eds) The Citrus industry. Vol. I. History, world distribution, botany, and varieties. Univ of California, Div of Agricultural Sci, Berkeley, pp 190–430
Swofford DL (2002) PAUP. Phylogenetic analysis using parsimony (* and other methods). Version 4.0. Sinauer Associates, Boston
Tanaka T (1954) Species problem in Citrus. A critical study of wild and cultivated units of Citrus, based upon field studies in their native homes. Japanese Society for the Promotion of Science, Ueno
Tanaka T (1977) Fundamental discussion of Citrus classification. Stud Citrol 14:1–6
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X Windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Torres AM, Soost RK, Diedenhofen U (1978) Leaf isozymes as genetic markers in Citrus. Am J Bot 65:869–881
Webber HJ (1967) History and development of the Citrus industry. In: Reuther W, Webber HJ, Batchelor LD (eds) The Citrus industry. Vol. I. history world distribution, botany, and varieties. Univ of California, Div of Agricultural Sci, Berkeley, pp 1–39
White TJ, Bruns T, Lee S, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Shinsky JJ, White TJ (eds) PCR protocols: a guide to methods and applications. Academic Press, San Diego, pp 315–322
Acknowledgments
Special thanks are given to colleagues and institutions for providing Citrus accessions and technical assistance: Dr. Do Dinh Ca-Research Institute for Fruits and Vegetables (RIFAV) (Ha Noi), Ms Vo Thi Tuyet-Phu Qui’s Fruit Tree Center, (PQFC), Dr. Pham Van Chuong-Agricultural Science Institute of North-Central Vietnam (ASINC), MSc Doan Nhan Ai- Fruit Tree Research & Development Center of ThuaThien-Hue (FRDC-Hue), Dr. Nguyen Minh Chau, Dr. Nguyen Thi Thu Hong, Ir Pham Ngoc Lieu-Institute of Southern Fruit Research Institute (SOFRI).
Author information
Authors and Affiliations
Corresponding author
Additional information
Tina Kyndt and Tran Nhan Dung equally contributed to this work.
Rights and permissions
About this article
Cite this article
Kyndt, T., Dung, T.N., Goetghebeur, P. et al. Analysis of ITS of the rDNA to infer phylogenetic relationships among Vietnamese Citrus accessions. Genet Resour Crop Evol 57, 183–192 (2010). https://doi.org/10.1007/s10722-009-9460-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10722-009-9460-0