
Abstract
Glycosaminoglycans (GAGs) are major components of cartilage extracellular matrix (ECM), which play an important role in tissue homeostasis not only by providing mechanical load resistance, but also as signaling mediators of key cellular processes such as adhesion, migration, proliferation and differentiation. Specific GAG types as well as their disaccharide sulfation patterns can be predictive of the tissue maturation level but also of disease states such as osteoarthritis. In this work, we used a highly sensitive liquid chromatography-tandem mass spectrometry (LC-MS/MS) method to perform a comparative study in terms of temporal changes in GAG and disaccharide composition between tissues generated from human bone marrow- and synovial-derived mesenchymal stem/stromal cells (hBMSC/hSMSC) after chondrogenic differentiation under normoxic (21% O2) and hypoxic (5% O2) micromass cultures. The chondrogenic differentiation of hBMSC/hSMSC cultured under different oxygen tensions was assessed through aggregate size measurement, chondrogenic gene expression analysis and histological/immunofluorescence staining in comparison to human chondrocytes. For all the studied conditions, the compositional analysis demonstrated a notable increase in the average relative percentage of chondroitin sulfate (CS), the main GAG in cartilage composition, throughout MSC chondrogenic differentiation. Additionally, hypoxic culture conditions resulted in significantly different average GAG and CS disaccharide percentage compositions compared to the normoxic ones. However, such effect was considerably more evident for hBMSC-derived chondrogenic aggregates. In summary, the GAG profiles described here may provide new insights for the prediction of cartilage tissue differentiation/disease states and to characterize the quality of MSC-generated chondrocytes obtained under different oxygen tension culture conditions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Articular cartilage defects do not heal spontaneously mainly due to the avascular nature of the tissue combined with the scarcity of resident stem cells. Current surgical methods such as microfracture and mosaicplasty can relieve pain to some extent but fail to generate functional and phenotypically stable hyaline-like cartilage tissue [1]. Autologous chondrocyte implantation (ACI), a cell-based strategy, in which the chondrocytes are isolated from non-weight bearing areas of articular cartilage, expanded in vitro and then implanted into the cartilage defect site, have failed to generate stable hyaline cartilage with long-term functionality. The limited clinical outcomes of ACI result from the fact that chondrocytes gradually lose their phenotype and undergo dedifferentiation during in vitro expansion [2, 3].
Mesenchymal stem/stromal cells (MSCs) are a promising alternative to chondrocytes for cartilage regeneration strategies due to their ease of isolation, higher in vitro expansion rates, multilineage differentiation capacity and low immunogenicity [4]. MSCs have been successfully isolated from different tissues including bone marrow, adipose tissue, umbilical cord, periosteum and synovium. However, MSCs from different sources have been shown to differ considerably in chondrogenic potential. Moreover, MSCs obtained from bone marrow, synovium and periosteum have been reported as superior sources for chondrogenesis [5,6,7,8]. Additionally, several studies have reported a superior chondrogenic ability of cells derived form human synovial joint tissues when compared with MSCs derived from bone marrow or adipose tissue [9,10,11,12].
MSCs chondrogenic differentiation is usually performed in high-density pellet or micromass cell culture systems to provide a 3D environment attempting to recapitulate the condensation step of endochondral bone formation during embryonic development. Previous work has suggested that micromass culture systems generate MSC-based cartilage tissues more hyaline-like and less hypertrophic when compared to pellet cultures [13].
In vivo, articular cartilage tissue is under hypoxic conditions (1%–6% O2 tension, compared to atmospheric air) [14]. Therefore, aiming to provide a closer mimicry of the native articular cartilage niche, researchers have explored low oxygen tension conditions as a strategy to enhance MSC chondrogenic differentiation [15,16,17].
Glycosaminoglycans (GAGs) are linear, highly charged carbohydrates with a repeating disaccharide unit and are among the principal functional constituents of articular cartilage. According to the structure and sulfation level of the repeating disaccharide, GAGs can be generally divided into four classes: heparan sulfate (HS), chondroitin sulfate (CS), hyaluronic acid (HA) and keratan sulfate (KS). GAGs play a crucial role in articular cartilage homeostasis not only by providing mechanical resistance to compressive loads, but also due to their involvement in several signaling pathways regulating important biological processes such as cell adhesion, growth and differentiation [18,19,20]. Indeed, changes in GAG composition and structure have been associated with different cell differentiation stages and with cartilage diseases such as osteoarthritis [21]. Accordingly, Chanalaris and colleagues reported that HS proteoglycan synthesis is dysregulated in human osteoarthritic cartilage [22]. Moreover, Veraldi et al reported significant HS structural differences in pathologic cartilage samples from patients with osteochondromas and chondrosarcomas in comparison to healthy cartilage [23]. Therefore, due to their biological importance, GAG production is one the main outcomes used to evaluate chondrogenic differentiation and assess the quality of the engineered cartilage tissues produced. Nevertheless, the great majority of the GAG measurements reported in the literature were obtained using the dimethylmethylene blue (DMMB) assay and correspond to total sulfated GAG content, being unable to discriminate among the different GAG types. Moreover, only few studies have reported GAG disaccharide composition of engineered cartilage tissues using electrophoresis or high-performance liquid chromatography (HPLC) methods [24, 25]. These methods lack the sensitivity and accuracy of liquid chromatography tandem mass spectrometry (LC-MS/MS), which might be critical when evaluating the low amounts usually generated in microscale culture strategies. In fact, our lab previously developed a highly sensitive and selective LC-MS/MS approach with multiple reaction monitoring (MRM), which was able to provide the GAG disaccharide composition of different types of tissues and biological samples, including urine, cell cultures, cell-derived extracellular matrices and intervertebral disc [26,27,28,29]. Such method was also successfully used to identify changes in GAG and disaccharide composition after early mesoderm and endoderm lineage commitment of human embryonic stem cells (ESC) [30] and to study temporal changes in the GAG composition during MSC differentiation towards the hepatic lineage [31].
In this work, the chondrogenic differentiation of human bone marrow-derived MSC (hBMSC) and human synovial-derived MSC (hSMSC) under different oxygen tensions (normoxia (21% O2) and hypoxia (5% O2)) was evaluated by micromass diameter measurements, RT-qPCR analysis and by histological/immunofluorescence stainings. To the best of our knowledge, this is the first use of LC-MS/MS analysis to identify temporal changes in GAG and disaccharide composition during hBMSC/hSMSC chondrogenic differentiation under normoxic/hypoxic culture conditions.
Materials and methods
Materials and reagents
Acetic acid, 2-aminoacridone (AMAC) and sodium cyanoborohydrade (NaCNBH4) were purchased from Sigma-Aldrich (St. Louis, MO). Dimethyl sulfoxide (DMSO), methanol, ammonium acetate and water (all HPLC grade) were obtained from Fisher Scientific (Springfield, NJ). Recombinant Flavobacterial heparinase I, II, III and chondroitin lyase ABC from Proteus vulgaris were expressed in our laboratory using Escherichia coli strains. The 17 unsaturated disaccharide standards of HS, HA and CS were acquired from Iduron (Manchester, UK) and their structures are shown in Supplementary Table 1.
Human cell sources
Bone marrow aspirates (Male 36 years) were obtained from Instituto Português de Oncologia Francisco Gentil, Lisboa-Potugal and an additional sample of fresh unprocessed bone marrow sample (Male 24 years) was purchased from Lonza (Basel, Switzerland). Synovium aspirates from donors undertaking routine arthroscopic surgery with no history of joint disease (Male 22 years and male 28 years) were obtained from Centro Hospitalar de Lisboa Ocidental, E.P.E, Hospital São Francisco Xavier, Lisboa, Portugal. All human samples were obtained from healthy donors after written informed consent and with the approval of the Ethics Committee of the respective clinical institution. Human bone marrow-derived MSC (hBMSC) and human synovial-derived MSC (hSMSC) were isolated following protocols previously developed in our laboratory [32, 33]. hBMSC and hSMSC were cultured using Dulbecco’s Modified Eagle’s Medium (DMEM, Gibco, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS, Life Technologies) and 1% antibiotics (penicillin-streptomycin, Pen-strep, Gibco) and cryopreserved in liquid nitrogen tanks until usage. Human chondrocytes (HC) were obtained from CELL Applications, Inc. and cultured using high-glucose DMEM supplemented with 10% FBS, 1X MEM non-essential aminoacids (Sigma, St. Louis, MO), 0.4 mM L-Proline (Sigma), 0.2 mM L-Ascorbic acid (Sigma) and 1% Pen-strep. The cultures of all cell sources were maintained in an incubator at 37 °C/5%CO2 with a humidified atmosphere and only cells with passage number between 3 and 5 were used in this work.
MSC characterization
Undifferentiated hBMSC and hSMSC morphology was observed under a phase contrast/fluorescence microscope (Olympus IX51 Inverted Microscope: Olympus America Inc., Melville, NY). For fluorescence staining, culture medium was removed and cells were washed twice with PBS, fixed with 4% paraformaldehyde (PFA; Santa Cruz Biotechnology, Dallas, TX) solution (in PBS) for 30 min and permeabilized with 0.1% Triton X-100 (Sigma-Aldrich) for 10 min. Upon permeabilization, cells were incubated with Phalloidin-TRITC (dilution 1:250, 2 μg/mL, Sigma-Aldrich) for 45 min in the dark. Then, cells were washed twice with PBS, stained with 4,6-diamino-2-phenylindole (DAPI, 1.5 μg/mL, Sigma-Aldrich) for 5 min, washed again with PBS and imaged under fluorescence microscopy.
hBMSC and hSMSC were tested for the expression of specific cell surface markers previously defined as minimal criteria to identify human MSC [34], using a panel of phycoerythrin (PE)-conjugated mouse anti-human monoclonal antibodies CD14, CD19, CD34, CD45, CD73, CD90, CD105 and HLA-DR and appropriate isotype controls (Biolegend, San Diego, CA). Thus, cells were incubated with each antibody for 15 min protected from light at room temperature and fixed with 2% PFA. Samples were analyzed by flow cytometry in a FACSCalibur™ instrument (Becton Dickinson, NJ) for quantification of the expression of each cell surface marker. A minimum of 10,000 events was collected for each sample and the CellQuest™ software (Becton Dickinson, NJ) was used for data acquisition and analysis.
hBMSC and hSMSC capacity to differentiate towards the osteogenic, adipogenic and chondrogenic lineage was evaluated. Cells were plated on 12-well plates at 6000 cells/cm2 and cultured with DMEM+10%FBS + 1% Pen-strep. When 80% confluence was reached, osteogenic and adipogenic differentiation was induced using StemPro™ Osteogenesis Differentiation Kit (Gibco™, Thermo Fisher Scientific) and StemPro™ Adipogenesis Differentiation Kit (Gibco™, Thermo Fisher Scientific), respectively. For chondrogenic differentiation, cells were concentrated to a density of 107 cells/mL and plated as droplets of 10 μL on ultra-low attachment 24-well culture plates (Falcon BD Biosciences, Corning, NY), and incubated for 1.5 h under humidified atmosphere at 37 °C and 5%CO2 to promote aggregation. Afterwards, excess fluid was removed and differentiation was induced using a StemPro™ Chondrogenesis Differentiation Kit (Gibco™, Thermo Fisher Scientific). The differentiation protocols were conducted for 14 days and the culture medium was changed twice a week. After 14 days of multilineage differentiation, the culture medium was removed, cells were washed with PBS, fixed in 2% PFA for 20 min at room temperature and rinsed in PBS. To confirm osteogenic differentiation, cells were incubated with a 4% (v/v) Fast Violet solution (Sigma-Aldrich) and Naphtol AS-MX Phosphate Alkaline solution (Sigma-Aldrich) for 45 min in the dark at room temperature. Afterwards, cells were washed three times with miliQ ultrapure water and once with PBS, and Von Kossa staining was performed by incubation with a 2.5% (w/v) silver nitrate solution (Sigma-Aldrich) for 30 min (at room temperature protected from light) to assess the presence of calcium deposits. Adipogenic differentiation was evaluated by incubating the cells with a 0.3% (w/v) Oil-Red-O solution (Sigma-Aldrich, in isopropanol) for 1 h at room temperature to identify lipid accumulation. Chondrogenic differentiation was assessed by incubation of the cellular aggregates with a 1% (w/v) Alcian Blue 8GX solution (Sigma-Aldrich, in 0.1 N HCl) for 1 h at room temperature to detect sulfated proteoglycans deposition. Finally, upon completion of the different staining protocols, cells were washed twice with PBS, rinsed with distilled water and imaged with a light microscope (LEICA® DMI3000B).
Chondrogenic differentiation of human BMSC, SMSC and chondrocytes under different oxygen tensions (normoxia-21% O2 and hypoxia-5% O2)
Human BMSC, SMSC and chondrocytes were harvested, concentrated and droplets of 15 μL containing 1.5 × 105 cells were placed in each well of ultra-low attachment 24-well culture plates, followed by an incubation for 1.5 h at 37 °C and 5%CO2 to promote initial cell aggregation. Afterwards, the aggregates were submersed with chondrogenic medium consisting of high glucose DMEM (Thermo Fisher Scientific) with 100 nM dexamethasone (Sigma-Aldrich), 50 μg/mL ascorbic acid 2-phosphate (Sigma-Aldrich), 40 μg/mL L-Proline (Sigma-Aldrich), 1 mM sodium pyruvate (Gibco), ITS™+ Premix supplement (6.25 μg/mL bovine insulin; 6.25 μg/mL transferrin; 6.25 μg/mL selenous acid; 5.33 μg/mL linoleic acid; 1.25 μg/mL BSA, Corning), Pen-strep (100 U/mL penicillin; 100 μg/mL streptomycin) and 10 ng/mL TGF-β3 (R&D Systems) and the cultures were placed in incubators under normoxia (21% O2) or hypoxia (5% O2) conditions. The chondrogenic differentiation protocol was performed for 21 days and culture medium was changed twice a week.
Chondrogenic aggregate size measurements
After 21 days of chondrogenic differentiation, the aggregates generated from the different cell sources under normoxia/hypoxia were imaged in a phase contrast microscope (Olympus IX51 Inverted Microscope). The estimation of the micromass aggregate diameters was performed by measuring 30 individual aggregates per condition (one per image) using the ImageJ software (ImageJ 1.51f, National Institutes of Health, USA).
Histological and immunofluorescence analysis
The final chondrogenic aggregates (day 21) derived from hBMSC, hSMSC and chondrocytes cultured under normoxia/hypoxia were fixed with 4% PFA for 20 min and washed with PBS. Afterwards, the aggregates were included in Tissue-Tek® Optimal Cutting Temperature (O.C.T.) Compound (VWR), frozen in liquid nitrogen and stored at −80 °C. The OCT blocks were sliced into 10 μm sections using a microtome cryostat (Microm HM 505E Cryostat, GMI, MN) at −20 °C and mounted in glass slides. The slides were washed twice in PBS (5 min each wash) and then washed with 0.1 M glycine (Sigma-Aldrich) solution in PBS for 10 min at room temperature to remove PFA residues. Samples were permeabilized with 0.1% (v/v) Triton solution in PBS for 10 min and incubated with a blocking solution (10% FBS in TBST: 20 mM Tris-HCl pH 8.0 (Sigma-Aldrich), 150 mM NaCl (Sigma-Aldrich), 0.05% (v/v) Tween-20 (Sigma-Aldrich)) for 30 min at room temperature and dried with a tissue.
For histological evaluation of the chondrogenic aggregates, the cross-sections were incubated with a 1% (w/v) Alcian Blue solution (in 0.1 N HCl) for 1 h and with a 0.1% (w/v) aqueous Safranin-O (Sigma-Aldrich) solution for 30 min to assess for the presence of sulfated GAGs. Then, the slides were washed three times with PBS, rinsed with distilled water and mounted with Mowiol mounting medium (Sigma-Aldrich). Images of the histological stainings of the chondrogenic aggregates were obtained with a light microscope (LEICA® DMI3000B).
For immunofluorescence analysis of the chondrogenic aggregates, the slides were incubated with primary antibodies (in blocking solution) for collagen II (1:200, mouse collagen II monoclonal antibody 6B3, ThermoFisher Scientific), aggrecan (1:400, mouse aggrecan monoclonal antibody BC-3, ThermoFisher Scientific) and lubricin (1:200, rabbit lubricin polyclonal antibody, ThermoFisher Scientific) overnight at 4 °C. Afterwards, the slides were washed three times with TBST (5 min each wash) and incubated with secondary antibodies Goat anti-mouse IgG- AlexaFluor 546 (1:500, ThermoFisher Scientific; for collagen II and aggrecan) and Goat anti-rabbit IgG- AlexaFluor 546 (1:500, ThermoFisher Scientific; for lubricin) for 45 min in the dark at room temperature. The slides were then washed with TBST (3 washes, 5 min each) and counterstained with DAPI for 5 min at room temperature. After washing the slides again with TBST, the samples were mounted with Mowiol. The slides containing the chondrogenic aggregates were examined under a confocal fluorescence microscope (Zeiss LSM 710).
RNA extraction and gene expression analysis by quantitative real time PCR
Total RNA was extracted from the final chondrogenic aggregates (day 21) derived from the different cell sources under normoxia/hypoxia as well as from the undifferentiated cells (day 0) using the RNeasy Mini kit (Quiagen, Hilden, Germany) according to the manufacturer’s guidelines and quantified using a Nanodrop (ND-100 Spectrophotometer, Nanodrop Technologies). cDNA was synthesized from the purified RNA using iScript™ Reverse Transcription Supermix (Bio-Rad, Hercules, CA) following the manufacturer’s guidelines. The real time quantitative PCR (RT-qPCR) analysis was performed using the TaqMan® Fast Advanced Master Mix (Applied Biosystems) and StepOnePlus real-time PCR system (Applied Biosystems). Reactions were run in triplicate using TaqMan® Gene Expression Assays (20X) (Thermo Fisher Scientific) for human Sox9 (Hs00165814_m1), human ACAN (Hs00153936_m1) and human GAPDH (Hs02758991_g1). The obtained CT values were normalized against the expression of housekeeping gene GAPDH and the analysis was performed using the 2-∆∆Ct method. Results for Sox9 and ACAN expressions in the hBMSC/hSMSC/HC-based chondrogenic aggregates cultured under normoxia/hypoxia are presented as fold-change expression levels relative to hBMSC/hSMSC/HC at day 0, respectively.
GAG disaccharide compositional analysis: Sample preparation, labeling and LC-MS/MS method
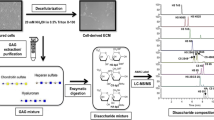
An overview of the general steps required for the LC-MS/MS GAG compositional analysis of the different chondrogenic micromass samples is presented in Fig. 1. Undifferentiated cells (day 0) and chondrogenic aggregates at different timepoints (days 7, 14 and 21 for hBMSC/hSMSC and day 21 for HC) cultured under normoxia/hypoxia were collected, incubated with the BugBuster 10X Protein Extraction Reagent (Millipore Sigma, MA) and sonicated in a bath containing ice for aggregate dissociation. Samples were desalted using a 3KDa molecular weight cutoff spin column (Millipore, MA) and washed thrice with distilled water. Afterwards, the columns were placed in new casing tubes and 300 μL of digestion buffer (50 mM ammonium acetate containing 2 mM calcium chloride, pH 7.0) was added to the filter unit. Recombinant heparin lyases I, II, III and recombinant chondroitin lyase ABC (10 mU each enzyme) were added to each sample and mixed well by pipetting. GAG enzymatic digestion was conducted by incubation overnight at 37 °C and terminated by centrifugation to remove the enzymes. The filter unit was washed twice with distilled water and the obtained filtrates containing the GAG disaccharides were lyophilized and stored at −20 °C until AMAC-labeling.

Experimental scheme of the steps for sample preparation for GAG disaccharide compositional analysis. GAGs from undifferentiated cells (hBMSC, hSMSC and chondrocytes) and respective derived aggregates undergoing chondrogenic differentiation in normoxic (21% O2) / hypoxic (5% O2) conditions were purified and digested by enzymes (heparinases and chondroitinase ABC), originating disaccharide mixtures. The disaccharide samples were then AMAC-labeled and analyzed by LC-MS/MS by comparison with external disaccharide standards
The dried disaccharide samples were labeled with 10 μL of 0.1 M AMAC in DMSO/acetic acid (17/3, V/V) solution by incubating for 10 min at room temperature, followed by the addition of 10 μL of 1 M aqueous NaCNBH4 solution and incubation for 1 h at 45 °C. A solution containing all 17 disaccharide standards (Supplementary Table 1) was prepared at a concentration of 0.5 ng/μL, labeled with AMAC and used for each run as an external standard. This external standard was prepared in the AMAC labeling solution, same as the biological samples, to minimize possible matrix suppression or enhancement effects. Upon termination of the AMAC-labeling reaction, samples were centrifuged and the respective supernatants were collected.
Disaccharide compositional analysis was done following a method previously developed in our laboratory [28]. LC was carry out on an Agilent 1200 LC system at 45 °C using an Agilent Poroshell 120 ECC18 (2.7 μm, 3.0 × 50 mm) column, 50 mM ammonium acetate aqueous solution as mobile phase A and methanol as mobile phase B. A flow rate of 300 μL/min was used to pass the mobile phases through the column. The gradient selected was: 0–10 min, 5–45% B; 10–10.2 min, 45–100%B; 10.2-14 min, 100%B; 14-22 min, 100–5%B; and the injection volume was 5 μL. The detector used consisted in a triple quadrupole mass spectrometry system equipped with an ESI source (Thermo Fisher Scientific, San Jose, CA). The online MS analysis was done in the MRM mode. The collected data was analyzed using the Thermo Xcalibur™ software (Thermo Fisher Scientific, San Jose, CA). GAG disaccharides present in the different samples were quantified by comparing the sample peak areas in the spectra to those of the external standards.
Statistical analysis
Results are presented as mean values ± standard deviation (SD) of three biological replicates for each of the two independent donors, unless specified differently. The statistical analysis of the LC-MS/MS data was performed using one-way ANOVA for multiple comparisons, followed by Tukey post-hoc test. Comparisons between gene expressions of the chondrogenic aggregates (from the same cell source) generated under 5% O2 and 21% O2 tensions were determined by the non-parametric Mann-Whitney U test. GraphPad Prism version 7 software was used in the analysis and data was considered to be significant when p-values obtained were less than 0.05 (95% confidence intervals, *p < 0.05).
Results
hBMSC and hSMSC characterization
The cell sources used in this study were characterized in terms of their morphology (Fig. 2A), immunophenotype (Fig. 2B) and multilineage differentiation capacity (Fig. 2C). Both hBMSC and hSMSC presented a standard morphological MSC phenotype with a long, fibroblastic appearance with defined nuclei and cytoskeleton. Regarding the immunophenotypical characterization, for both sources, less than 2% of the population expressed hematopoietic lineage markers CD14, CD19, CD34, CD45 and HLA-DR. Considering the positive markers (CD73, CD90 and CD105), the expression of CD73 and CD105 was above to 95% for both hBMSC and hSMSC. In the case of CD90, while more than 98% of hBMSC expressed this marker, hSMSC presented a expression of approximately 82%. In terms of the in vitro multilineage differentiation potential, after 2 weeks of induction, both hBMSC and hSMSC were able to differentiate towards osteogenic, adipogenic and chondrogenic lineages as confirmed by ALP/Von Kossa, Oil Red-O and Alcian Blue staining, respectively.

Characterization of hBMSC and hSMSC used in this study. The morphology of hBMSC and hSMSC was observed by light and fluorescence microscopy after DAPI/Phalloidin staining (DAPI stains cell nuclei blue and Phalloidin stains actin-rich cell cytoskeleton red (A). Immunophenotypical analysis of hBMSC and hSMSC (B): both sources express MSC characteristic markers CD73, CD90 and CD105 and present low expression levels (<2%) of CD45, CD34, CD14, CD19 and HLA-DR. Multilineage differentiation potential of hBMSC and hSMSC assessed after 14 days under osteogenic, adipogenic and chondrogenic induction (C). Osteogenic differentiation was confirmed by ALP/Von Kossa staining. Adipogenesis was evaluated by staining the cells with Oil Red-O. Chondrogenic differentiation was assessed by Alcian Blue staining. Values are represented as mean ± SD of two independent donors for each cell source. Scale bars: 100 μm
Evaluation of the chondrogenic differentiation of human BMSC, SMSC and chondrocytes under normoxia (21% O2) / hypoxia (5% O2)
At the end of the chondrogenic differentiation protocol (day 21), the final hBMSC/hSMSC/HC-based chondrogenic micromass tissues generated under normoxia (21% O2) and hypoxia (5% O2) were evaluated in terms of typical cartilage ECM proteins/proteoglycans expression (Fig. 3), the aggregate diameter (Fig. 4) and chondrogenic marker genes expression (Fig. 5).

Histological (Alcian Blue and Safranin-O stainings) and immunofluorescence (Collagen II, Aggrecan and Lubricin) analysis of the final (day 21) chondrogenic aggregates generated by hBMSC, hSMSC and HC under normoxia (21% O2)/hypoxia (5% O2). The immunofluorescence samples were counterstained with DAPI. Scale bars: 100 μm

Average diameter and respective diameter distribution of the aggregates generated by hBMSC, hSMSC and HC after 21 days of micromass chondrogenic culture under normoxia (21% O2)/hypoxia (5% O2). Data are presented as mean ± SD, n = 30 individual aggregates

RT-qPCR analysis of the final (day 21) chondrogenic aggregates generated by hBMSC, hSMSC and HC under normoxia (21% O2)/hypoxia (5% O2). Sox 9 and ACAN gene expressions are normalized against the housekeeping gene GAPDH and presented as fold-change levels relative to the respective cell source (hBMSC, hSMSC and HC) at day 0. Data are presented as mean ± SD, n = 3. *p < 0.05
Regardless of the cell source and the oxygen tension used, all the chondrogenic micromass tissues stained positively for the presence of GAGs after Alcian Blue and Safranin-O stainings. Additionally, immunofluorescence analysis showed that all the produced tissues stained positive for the presence of typical cartilage ECM components collagen II, aggrecan and lubricin (Fig. 3).
As it is possible to observe in Fig. 4, for all the cell sources, hypoxic cultures lead to the formation of aggregates with higher average diameters than the ones obtained under normoxia. Considering the MSC sources, within the same oxygen tension, hSMSC-derived chondrogenic aggregates presented higher average diameters than hBMSC-derived ones.
Figure 5 shows the RT-qPCR analysis performed in the final cartilage engineered tissues generated from hBMSC, hSMSC and HC under normoxia (21% O2) and hypoxia (5% O2). Although all the conditions showed upregulation of Sox9 and ACAN chondrogenic marker genes, tissues obtained from different MSC sources showed different responses to low oxygen tensions. While for hBMSC-derived chondrogenic aggregates, hypoxic cultures presented significantly higher Sox9 and ACAN expressions than normoxic ones, an opposite trend was observed for hSMSC-derived chondrogenic aggregates. HC-derived aggregates cultured under hypoxia also showed higher chondrogenic gene expressions than normoxic cultures, however significant differences were only observed for ACAN.
GAG and respective disaccharide changes during human BMSC, SMSC and chondrocyte chondrogenic differentiation under normoxia (21% O2) / hypoxia (5% O2)
GAG remodeling during the chondrogenic differentiation of hBMSC and hSMSC under hypoxic/normoxic culture conditions was studied using LC-MS/MS analysis. For that, samples were harvested at different timepoints during the differentiation (days 0, 7, 14 and 21) and GAGs were purified, enzymatically digested and labeled for compositional disaccharide analysis (Fig. 1). HC-aggregates differentiated under the same conditions were used as controls. In Fig. 6 it is shown the temporal changes in the average percentage GAG composition of hBMSC- and hSMSC-derived chondrogenic aggregates throughout differentiation. Significantly distinct GAG average compositions were observed among the different cell sources. In their undifferentiated state (day 0), hBMSC were mainly composed by HA (74%, of total GAG content) with lower percentages of CS (14%) and HS (12%). In contrast, the main GAG component in undifferentiated hSMSC was CS (55%) with lower average percentages of HA (23%) and HS (22%), which was more similar to HC average GAG composition (CS: 58%, HS: 33% and HA: 9%) than hBMSC. During chondrogenic differentiation, for all the conditions tested, it is possible to observe an increase over time of CS average percentages and corresponding lower average percentages of HS and HA. Hypoxia conditions appeared to favor significantly increased CS average percentages compared to normoxia in hBMSC-derived chondrogenic aggregates. However, the same trend was not observed for the cartilage tissues derived from other cell sources. In fact, at the end of the differentiation protocol (day 21), hBMSC-derived chondrogenic aggregates obtained under hypoxic conditions showed significantly higher CS and reduced HA, HS average percentages than the ones generated at normoxic conditions (CS: 91%, HA: 8% and HS: 1% at hypoxia vs. CS: 59%, HA: 30% and HS: 11% at normoxia). In contrast, while the GAG composition for hSMSC-derived cartilage tissues obtained under hypoxia was 71% CS, 25% HA and 4% HS, tissues generated under normoxia showed a GAG composition of 86% CS, 9% HA and 5% HS. In addition, HC-derived cartilage tissues were mainly composed by CS with higher average percentages than the ones observed for the other cell sources (CS: 95% at hypoxia and 94% at normoxia) and no significant differences in GAG composition were noticed between HC-aggregates generated under different oxygen tensions.

Average percentage GAG composition of undifferentiated cells (day 0) and during chondrogenic differentiation (days 7, 14 and 21) micromass culture of hBMSC and hSMSC under normoxic (21% O2)/hypoxic (5% O2) conditions. HC were used for comparison at days 0 and 21. Data are presented as mean ± SD of three replicates for each donor (n = 6) for hBMSC and hSMSC, and n = 3 for HC. *p < 0.05
The average percentage HS and CS disaccharide compositional changes during the chondrogenic differentiation of hBMSC, hSMSC and HC under hypoxia/normoxia are presented in Fig. 7 and Fig. 8, respectively. Regarding HS disaccharides, all the samples were mainly composed by HS 0S, with lower percentages of HS NS and HS NS2S. Generally, during chondrogenic differentiation, the average percentages for the different HS disaccharides were nearly maintained and changes resulting from culture under different oxygen tensions were not noticed. However, some significant differences were observed in the HS composition of the different cell sources, mainly at the end of the protocol (day 21). At day 21, HC-derived aggregates showed significantly higher average percentages of HS 0S than the hSMSC-derived ones, contrarily to what was observed in these cells undifferentiated state (day 0).

Average percentage HS disaccharide composition of undifferentiated cells (day 0) and during chondrogenic differentiation (days 7, 14 and 21) micromass culture of hBMSC and hSMSC under normoxic (21% O2)/hypoxic (5% O2) conditions. HC were used for comparison at days 0 and 21. Data are presented as mean ± SD of three replicates for each donor (n = 6) for hBMSC and hSMSC,and n = 3 for HC. *p < 0.05

Average percentage CS disaccharide composition of undifferentiated cells (day 0) and during chondrogenic differentiation (days 7, 14 and 21) micromass culture of hBMSC and hSMSC under normoxic (21% O2)/hypoxic (5% O2) conditions. HC were used for comparison at days 0 and 21. Data are presented as mean ± SD of three replicates for each donor (n = 6) for hBMSC and hSMSC, and n = 3 for HC. *p < 0.05
In Fig. 8 it is possible to observe that, for all the conditions, CS was mainly composed by CS 4S and CS 6S with lower percentages of CS 0S. Significant differences in CS 4S and CS 6S average percentages were observed between the tissues generated from different cell sources and also as a result of culture under different oxygen tensions. While in their undifferentiated state (day 0), all cell types presented higher average percentages for CS 4S than CS 6S, after 21 days of chondrogenic differentiation, the obtained tissues (with the exception of hBMSC-derived cartilaginous tissue) showed higher amounts of CS 6S than CS 4S. Moreover, for tissues derived from all cell sources, this increase in CS 6S average percentages (and subsequent decrease in CS 4S) was clearly favored by hypoxia. Noteworthy, HC-derived cartilage tissues presented significantly higher CS 6S (and lower CS 4S) average percentages than the tissues obtained from both MSC sources.
Discussion
To the best of our knowledge, this study represents one of the first reports on the use of highly sensitive and selective LC-MS/MS methods to evaluate the GAG remodeling during MSC chondrogenic differentiation. Herein, we compared the GAG composition of tissues generated from two different human MSC sources (hBMSC and hSMSC) when cultured under different oxygen tensions (normoxia-21% O2 and hypoxia-5% O2). HC were cultured under the same conditions as the other cell sources and used throughout this study as control samples. Both sources used in this work were characterized and proved to be compliant with the criteria defined by Dominici et al for MSC identification [34]. Regarding the immunophenotypic analysis, hSMSC showed slightly decreased expression of CD90 (<95%). However, since the panel of markers proposed by Dominici et al focused on the identification of hBMSC, there are no specific defined sets of markers to identify MSC isolated from other sources, which might have its own intrinsic levels of markers expression. Moreover, such decreased CD90 expression in hSMSC was previously described in other studies [35, 36].
The chondrogenic aggregates derived from hSMSC presented higher average diameters that the ones derived from hBMSC, regardless of the oxygen tension used (1.4-fold in 21% O2 and 1.3-fold in 5% O2). In accordance with our results, Ogata and colleagues obtained MSC-derived tissues with the same range of millimeter scale diameters and showed that hSMSC-derived tissues presented diameters 1.2-fold larger than hBMSC-derived tissues [12]. For all the cell sources, chondrogenic aggregates produced under hypoxia showed higher diameters (1.3-fold for hBMSC, 1.1-fold for hSMSC and 1.5-fold for HC) than the ones generated at atmospheric oxygen tension. This hypoxia-induced increase in aggregate size was also reported for hBMSC-derived micropellet tissues produced under 2% O2 [37]. However, in contrast to what we showed, Bae and colleagues did not observe any considerable differences between the diameter of the hSMSC-derived pellets obtained under 21% O2 and 5% O2 [17].
RT-qPCR analysis showed increased ACAN and Sox9 expressions for hBMSC- and HC-derived cartilage tissues when cultured under hypoxia, which is concordant with previous studies [16, 37, 38]. In contrast with the other cell sources and with the reported by Bae and colleagues for hSMSC-derived pellets, the final hSMSC-derived cartilage tissues obtained under hypoxia showed lower expressions of ACAN and Sox9 than the ones generated at normoxic conditions [17]. Nevertheless, contrarily to hBMSC and HC, the effect of low oxygen-cultures in hSMSC chondrogenesis is not fully characterized yet as only very few studies have addressed this issue. Additionally, previous studies reported donor-dependency in the response of hBMSC-derived pellets to hypoxic cultures by showing that chondrogenic marker genes were differently regulated among different donors, which would probably also apply to other MSC sources [15, 39]. The role of hypoxia during MSC chondrogenesis is not fully understood yet and despite hypoxia has been generally associated with enhanced chondrogenic differentiation, some incongruent results have been reported [40]. In fact, Cicione and colleagues reported inhibited Sox9 and ACAN chondrogenic genes expression in hBMSC pellets cultured under hypoxia in comparison to the ones produced under normoxic conditions [41]. However, the comparison between different studies is limited due to MSCs heterogeneity, differences in chondrogenic differentiation protocols and various oxygen tensions used. Therefore, the development of standardized protocols for hypoxic cultures might contribute to a broader consensus on the effects of hypoxia in the chondrogenic differentiation of MSCs.
LC-MS/MS analysis revealed significant changes in GAG and disaccharide composition during hBMSC, hSMSC and HC chondrogenic differentiation under normoxic/hypoxic conditions. Undifferentiated hSMSC presented a GAG composition profile much more similar to HC than hBMSC. This might be related with the fact that hSMSC are described as more prone for chondrocyte differentiation than hBMSC. In fact, it was previously shown that the gene expression profiles of hSMSC and chondrocytes are closer to each other than those of extra-articular tissue-derived MSC, including hBMSC [42]. As the most predominant GAG in articular cartilage is CS, variations in CS relative amounts might provide insights about the differentiation state of MSC-derived tissues produced. In our analysis, we observed an increase in the CS average percentages during the chondrogenic differentiation of both hBMSC and hSMSC. Additionally, we observed that hypoxia affected differently the GAG remodeling of hBMSC and hSMSC. While for hBMSC-derived cartilage tissues, hypoxia resulted in higher average percentages of CS than normoxia, an opposite trend was observed for hSMSC-derived cartilage tissues. In fact, these different hypoxia-induced changes in CS composition of the tissues generated by hBMSC and hSMSC are coherent and might be related with the trends observed for ACAN gene expression.
The GAG content of HC-derived cartilage tissues was mainly composed by CS (95% at 5% O2 and 94% at 21% O2) and was not significantly affected by oxygen tension. These percentages are similar to the values reported by Osago et al (ranging from 95.2–96.3%, depending on the tissue digestion method used) for the total CS composition of porcine articular cartilage analyzed by LC-MS/MS [43]. The lower CS average percentages observed for the MSC-derived cartilage tissues might suggest early differentiation states. Nevertheless, the CS percentage values observed for hBMSC-derived cartilage tissues at 5% O2 (91%) and hSMSC-derived cartilage tissues at 21% O2 (86%) are relatively close to the ones verified for HC-derived cartilage tissues.
The disaccharide composition of CS in articular cartilage, particularly the CS 6S/CS 4S ratio is known to vary with age and degeneration of the tissue [44]. In fact, while during embryonic development CS chains are exclusively CS 6S, they change to be equally composed by CS 4S and CS 6S from fetal development to adolescence, and composed by more CS 6S than CS 4S in adult cartilage [45, 46]. Additionally, the sulfation pattern of CS in human osteoarthritic cartilage has been shown to consist primarily of CS 6S with lower levels of CS 4S [47, 48]. Therefore, changes in the sulfation patterns, namely the relations between CS 6S and CS 4S percentages observed during hBMSC/hSMSC chondrogenic differentiation could provide valuable insights about the maturation level of the tissues generated. With the exception of hBMSC-derived cartilage tissue at 21% O2, all the other samples showed higher CS 6S/lower CS 4S percentages (higher CS 6S/CS 4S ratio) in relation to the respective cell source at day 0. HC-derived cartilage tissues presented higher CS 6S/CS 4S ratios than MSC-derived tissues, which might suggest a higher tissue maturation level. Additionally, hypoxia demonstrated to have a significant effect in the sulfation pattern of the final tissues, as for all the cell sources, tissues produced under 5% O2 presented higher CS 6S/CS 4S ratios than the ones generated at 21% O2. These findings are concordant with the studies reporting that hypoxia enhance MSC chondrogenesis towards more mature cartilage tissues [16]. Concerning cartilage regeneration strategies, besides oxygen tension and cell source, the scaffold material has also been shown to affect the disaccharide composition of the cartilage tissue produced [25, 49].
In summary, we used a highly sensitive LC-MS/MS method to provide a novel analysis of the GAG remodeling during MSC chondrogenesis and assess how it varies with the MSC source and oxygen tension culture conditions. However, some limitations are important to highlight. This method was based on disaccharide analysis through the use of chondroitinase ABC and heparinases, so, it could only detect CS, HS and HA. Therefore, additional methodological developments should be pursued in order to allow for the quantification of KS, which is known to be present in articular cartilage. As this method do not assess core proteins, it would be interesting to perform this analysis in combination with a proteomics approach in order to provide better information about the composition and functionality of the final in vitro produced tissues. A detailed analysis of the GAG remodeling during chondrogenesis is important not only to better understand the mechanisms of cartilage development and disease, but also to provide new insights for improved cartilage regeneration strategies and new methods to characterize the quality of the tissue substitutes produced.
References
Richter, D.L., Schenck, R.C., Wascher, D.C., Treme, G.: Knee articular cartilage repair and restoration techniques: a review of the literature. Sports Health. 8, 153–160 (2016). https://doi.org/10.1177/1941738115611350
Darling, E.M., Athanasiou, K.A.: Rapid phenotypic changes in passaged articular chondrocyte subpopulations. J. Orthop. Res. 23, 425–432 (2005). https://doi.org/10.1016/j.orthres.2004.08.008
Rackwitz, L., Djouad, F., Janjanin, S., Nöth, U., Tuan, R.S.: Functional cartilage repair capacity of de-differentiated, chondrocyte- and mesenchymal stem cell-laden hydrogels in vitro. Osteoarthr. Cartil. 22, 1148–1157 (2014). https://doi.org/10.1016/j.joca.2014.05.019
Huang, Y.Z.S., Xie, H.Q., Silini, A., Parolini, O., Zhang, Y., Deng, L., Huang, Y.C.: Mesenchymal stem/progenitor cells derived from articular cartilage. Synovial Membrane and Synovial Fluid for Cartilage Regeneration: Current Status and Future Perspectives. Stem Cell Rev. Reports. 13, 575–586 (2017). https://doi.org/10.1007/s12015-017-9753-1
Li, C.Y., Wu, X.Y., Tong, J.B., Yang, X.X., Zhao, J.L., Zheng, Q.F., Zhao, G.B., Ma, Z.J.: Comparative analysis of human mesenchymal stem cells from bone marrow and adipose tissue under xeno-free conditions for cell therapy. Stem Cell Res. Ther. 6, 55 (2015). https://doi.org/10.1186/s13287-015-0066-5
Tan, A.R., Hung, C.T.: Concise review: Mesenchymal stem cells for functional cartilage tissue engineering: taking cues from chondrocyte-based constructs. Stem Cells Transl. Med. 6, 1295–1303 (2017). https://doi.org/10.1002/sctm.16-0271
Yoshimura, H., Muneta, T., Nimura, A., Yokoyama, A., Koga, H., Sekiya, I.: Comparison of rat mesenchymal stem cells derived from bone marrow, synovium, periosteum, adipose tissue, and muscle. Cell Tissue Res. 327, 449–462 (2007). https://doi.org/10.1007/s00441-006-0308-z
Bernardo, M.E., Emons, J.A.M., Karperien, M., Nauta, A.J., Willemze, R., Roelofs, H., Romeo, S., Marchini, A., Rappold, G.A., Vukicevic, S., Locatelli, F., Fibbe, W.E.: Human mesenchymal stem cells derived from bone marrow display a better chondrogenic differentiation compared with other sources. Connect. Tissue Res. 48, 132–140 (2007). https://doi.org/10.1080/03008200701228464
Sakaguchi, Y., Sekiya, I., Yagishita, K., Muneta, T.: Comparison of human stem cells derived from various mesenchymal tissues: superiority of synovium as a cell source. Arthritis Rheum. 52, 2521–2529 (2005). https://doi.org/10.1002/art.21212
Shirasawa, S., Sekiya, I., Sakaguchi, Y., Yagishita, K., Ichinose, S., Muneta, T.: In vitro chondrogenesis of human synovium-derived mesenchymal stem cells: optimal condition and comparison with bone marrow-derived cells. J. Cell. Biochem. 97, 84–97 (2006). https://doi.org/10.1002/jcb.20546
Fan, J., Varshney, R.R., Ren, L., Cai, D., Wang, D.A.: Synovium-derived mesenchymal stem cells: a new cell source for musculoskeletal regeneration. Tissue Eng. Part B. Rev. 15, 75–86 (2009). https://doi.org/10.1089/ten.teb.2008.0586
Ogata, Y., Mabuchi, Y., Yoshida, M., Suto, E.G., Suzuki, N., Muneta, T., Sekiya, I., Akazawa, C.: Purified human synovium mesenchymal stem cells as a good resource for cartilage regeneration. PLoS One. 10, e0129096 (2015). https://doi.org/10.1371/journal.pone.0129096
Zhang, L., Su, P., Xu, C., Yang, J., Yu, W., Huang, D.: Chondrogenic differentiation of human mesenchymal stem cells: a comparison between micromass and pellet culture systems. Biotechnol. Lett. 32, 1339–1346 (2010). https://doi.org/10.1007/s10529-010-0293-x
Zhou, S., Cui, Z., Urban, J.P.G.: Factors influencing the oxygen concentration gradient from the synovial surface of articular cartilage to the cartilage-bone interface: a modeling study. Arthritis Rheum. 50, 3915–3924 (2004). https://doi.org/10.1002/art.20675
Adesida, A.B., Mulet-Sierra, A., Jomha, N.M.: Hypoxia mediated isolation and expansion enhances the chondrogenic capacity of bone marrow mesenchymal stromal cells. Stem Cell Res Ther. 3, 9 (2012). https://doi.org/10.1186/scrt10022385573
Leijten, J., Georgi, N., Moreira Teixeira, L., van Blitterswijk, C.A., Post, J.N., Karperien, M.: Metabolic programming of mesenchymal stromal cells by oxygen tension directs chondrogenic cell fate. Proc. Natl. Acad. Sci. 111, 13954–13959 (2014). https://doi.org/10.1073/pnas.1410977111
Bae, H.C., Park, H.J., Wang, S.Y., Yang, H.R., Lee, M.C., Han, H.-S.: Hypoxic condition enhances chondrogenesis in synovium-derived mesenchymal stem cells. Biomater. Res. 22, 1–8 (2018). https://doi.org/10.1186/s40824-018-0134-x
Weyers, A., Linhardt, R.J.: Neoproteoglycans in tissue engineering. FEBS J. 280, 2511–2522 (2013). https://doi.org/10.1111/febs.12187
Gasimli, L., Linhardt, R.J., Dordick, J.S.: Proteoglycans in stem cells. Biotechnol. Appl. Biochem. 59, 65–76 (2012). https://doi.org/10.1002/bab.1002
Uygun, B.E., Stojsih, S.E., Matthew, H.W.T.: Effects of immobilized glycosaminoglycans on the proliferation and differentiation of mesenchymal stem cells. Tissue Eng. Part A. 15, 3499–3512 (2009). https://doi.org/10.1089/ten.TEA.2008.0405
Plaas, A.H.K., West, L.A., Wong-Palms, S., Nelson, F.R.T.: Glycosaminoglycan sulfation in human osteoarthritis: disease-related alterations at the non-reducing termini of chondroitin and dermatan sulfate. J. Biol. Chem. 273, 12642–12649 (1998). https://doi.org/10.1074/jbc.273.20.12642
Chanalaris, A., Clarke, H., Guimond, S.E., Vincent, T.L., Turnbull, J.E., Troeberg, L.: Heparan sulfate proteoglycan synthesis is Dysregulated in human osteoarthritic cartilage. Am. J. Pathol. 189, 632–647 (2019). https://doi.org/10.1016/j.ajpath.2018.11.011
Veraldi, N., Parra, A., Urso, E., Cosentino, C., Locatelli, M., Corsini, S., Pedrini, E., Naggi, A., Bisio, A., Sangiorgi, L.: Structural features of heparan sulfate from multiple osteochondromas and chondrosarcomas. Molecules. 23, 3277 (2018). https://doi.org/10.3390/molecules23123277
Wan, S., Borland, S., Richardson, S.M., Merry, C.L.R., Saiani, A., Gough, J.E.: Self-assembling peptide hydrogel for intervertebral disc tissue engineering. Acta Biomater. 46, 29–40 (2016). https://doi.org/10.1016/j.actbio.2016.09.033
Mouw, J.K., Case, N.D., Guldberg, R.E., Plaas, A.H.K., Levenston, M.E.: Variations in matrix composition and GAG fine structure among scaffolds for cartilage tissue engineering. Osteoarthr. Cartil. 13, 828–836 (2005). https://doi.org/10.1016/j.joca.2005.04.020
Li, G., Li, L., Tian, F., Zhang, L., Xue, C., Linhardt, R.J.: Glycosaminoglycanomics of cultured cells using a rapid and sensitive LC-MS/MS approach. ACS Chem. Biol. 10, 1303–1310 (2015)
Liu, X., Krishnamoorthy, D., Lin, L., Xue, P., Zhang, F., Chi, L., Linhardt, R.J., Iatridis, J.C.: A method for characterising human intervertebral disc glycosaminoglycan disaccharides using liquid chromatography-mass spectrometry with multiple reaction monitoring. Eur. Cell. Mater. 35, 117–131 (2018). https://doi.org/10.22203/eCM.v035a09
Sun, X., Li, L., Overdier, K.H., Ammons, L.A., Douglas, I.S., Burlew, C.C., Zhang, F., Schmidt, E.P., Chi, L., Linhardt, R.J.: Analysis of Total human urinary glycosaminoglycan disaccharides by liquid chromatography-tandem mass spectrometry. Anal. Chem. 87, 6220–6227 (2015). https://doi.org/10.1021/acs.analchem.5b00913
Silva, J.C., Carvalho, M.S., Han, X., Xia, K., Mikael, P.E., Cabral, J.M.S., Ferreira, F.C., Linhardt, R.J.: Compositional and structural analysis of glycosaminoglycans in cell-derived extracellular matrices. Glycoconj. J. 36, 141–154 (2019). https://doi.org/10.1007/s10719-019-09858-2
Gasimli, L., Hickey, A.M., Yang, B., Li, G., Dela Rosa, M., Nairn, A.V., Kulik, M.J., Dordick, J.S., Moremen, K.W., Dalton, S., Linhardt, R.J.: Changes in glycosaminoglycan structure on differentiation of human embryonic stem cells towards mesoderm and endoderm lineages. Biochim. Biophys. Acta - Gen. Subj. 1840, 1993–2003 (2014). https://doi.org/10.1016/j.bbagen.2014.01.007
Mikael, P.E., Willard, C., Koyee, A., Barlao, C.-G., Liu, X., Han, X., Ouyang, Y., Xia, K., Linhardt, R.J., Dordick, J.S.: Remodeling of Glycosaminoglycans during differentiation of adult human bone Mesenchymal stromal cells toward hepatocytes. Stem Cells Dev. 28, 278–289 (2019). https://doi.org/10.1089/scd.2018.0197
Dos Santos, F., Andrade, P.Z., Boura, J.S., Abecasis, M.M., da Silva, C.L., Cabral, J.M.S.: Ex vivo expansion of human mesenchymal stem cells: a more effective cell proliferation kinetics and metabolism under hypoxia. J. Cell. Physiol. 223, 27–35 (2010). https://doi.org/10.1002/jcp.21987
Santhagunam, A., Dos Santos, F., Madeira, C., Salgueiro, J.B., Cabral, J.M.S.: Isolation and ex vivo expansion of synovial mesenchymal stromal cells for cartilage repair. Cytotherapy. 16, 440–453 (2013). https://doi.org/10.1016/j.jcyt.2013.10.010
Dominici, M., Le Blanc, K., Mueller, I., Slaper-Cortenbach, I., Marini, F., Krause, D., Deans, R., Keating, A., Prockop, D., Horwitz, E.: Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 8, 315–317 (2006). https://doi.org/10.1080/14653240600855905
Nagase, T., Muneta, T., Ju, Y.J., Hara, K., Morito, T., Koga, H., Nimura, A., Mochizuki, T., Sekiya, I.: Analysis of the chondrogenic potential of human synovial stem cells according to harvest site and culture parameters in knees with medial compartment osteoarthritis. Arthritis Rheum. 58, 1389–1398 (2008). https://doi.org/10.1002/art.23418
Ferro, T., Santhagunam, A., Madeira, C., Salgueiro, J.B., da Silva, C.L., Cabral, J.M.S.: Successful isolation and ex vivo expansion of human mesenchymal stem/stromal cells obtained from different synovial tissue-derived (biopsy) samples. J. Cell. Physiol. 234, 3973–3984 (2019). https://doi.org/10.1002/jcp.27202
Markway, B.D., Tan, G.K., Brooke, G., Hudson, J.E., Cooper-White, J.J., Doran, M.R.: Enhanced chondrogenic differentiation of human bone marrow-derived mesenchymal stem cells in low oxygen environment micropellet cultures. Cell Transplant. 19, 29–42 (2010). https://doi.org/10.3727/096368909X478560
Lafont, J.E., Talma, S., Hopfgarten, C., Murphy, C.L.: Hypoxia promotes the differentiated human articular chondrocyte phenotype through SOX9-dependent and -independent pathways. J. Biol. Chem. 283, 4778–4786 (2008). https://doi.org/10.1074/jbc.M707729200
Gawlitta, D., van Rijen, M.H.P., Schrijver, E.J.M., Alblas, J., Dhert, W.J.A.: Hypoxia impedes hypertrophic Chondrogenesis of human multipotent stromal cells. Tissue Eng. Part A. 18, 1957–1966 (2012). https://doi.org/10.1089/ten.tea.2011.0657
Shang, J., Liu, H., Li, J., Zhou, Y.: Roles of hypoxia during the Chondrogenic differentiation of Mesenchymal stem cells. Curr. Stem Cell Res. Ther. 9, 141–147 (2014). https://doi.org/10.2174/1574888x09666131230142459
Cicione, C., Muiños-López, E., Hermida-Gómez, T., Fuentes-Boquete, I., Díaz-Prado, S., Blanco, F.J.: Effects of Severe Hypoxia on Bone Marrow Mesenchymal Stem Cells Differentiation Potential. Stem Cells Int. 232896 (2013). https://doi.org/10.1155/2013/232896
Segawa, Y., Muneta, T., Makino, H., Nimura, A., Mochizuki, T., Ju, Y.J., Ezura, Y., Umezawa, A., Sekiya, I.: Mesenchymal stem cells derived from synovium, meniscus, anterior cruciate ligament, and articular chondrocytes share similar gene expression profiles. J. Orthop. Res. 27, 435–441 (2009). https://doi.org/10.1002/jor.20786
Osago, H., Kobayashi-Miura, M., Hamasaki, Y., Hara, N., Hiyoshi, M., Tsuchiya, M.: Complete solubilization of cartilage using the heat-stable protease thermolysin for comprehensive GAG analysis. Anal. Biochem. 548, 115–118 (2018). https://doi.org/10.1016/j.ab.2018.02.028
Lauder, R.M., Huckerby, T.N., Brown, G.M., Bayliss, M.T., Nieduszynski, I.A.: Age-related changes in the sulphation of the chondroitin sulphate linkage region from human articular cartilage aggrecan. Biochem. J. 358, 523–528 (2001). https://doi.org/10.1042/0264-6021:3580523
Sharma, A., Rees, D., Roberts, S., Kuiper, N.J.: A case study: glycosaminoglycan profiles of autologous chondrocyte implantation (ACI) tissue improve as the tissue matures. Knee. 24, 149–157 (2017). https://doi.org/10.1016/j.knee.2016.10.002
Hitchcock, A.M., Yates, K.E., Costello, C.E., Zaia, J.: Comparative glycomics of connective tissue glycosaminoglycans. Proteomics. 8, 1384–1397 (2008). https://doi.org/10.1002/pmic.200700787
Hitchcock, A.M., Yates, K.E., Shortkroff, S., Costello, C.E., Zaia, J.: Optimized extraction of glycosaminoglycans from normal and osteoarthritic cartilage for glycomics profiling. Glycobiology. 17, 25–35 (2007). https://doi.org/10.1093/glycob/cwl046
Lin, T.-S., Hsieh, C.-H., Kuo, C., Juang, Y.-P., Hsieh, Y.S., Chiang, H., Hung, S.-C., Jiang, C.-C., Liang, P.-H.: Sulfation pattern of chondroitin sulfate in human osteoarthritis cartilages reveals a lower level of Chondroitin-4-sulfate. Carbohydr. Polym. 229, 115496 (2020). https://doi.org/10.1016/j.carbpol.2019.115496
Wang, Q.G., Hughes, N., Cartmell, S.H., Kuiper, N.J.: The composition of hydrogels for cartilage tissue engineering can influence glycosaminoglycan profiles. Eur. Cells Mater. 19, 86–95 (2010). https://doi.org/10.22203/eCM.v019a09
Acknowledgements
This study was financed by Center for Biotechnology and Interdisciplinary Studies-Rensselaer Polytechnic Institute funds and by the National Institutes of Health though the Grant # DK111958. This work was also supported by funding received by iBB-Institute for Bioengineering and Biosciences through Programa Operacional Regional de Lisboa 2020 (Project N. 007317), through the EU COMPETE Program and from FCT-Portuguese Foundation for Science and Technology (Programme grant UID/BIO/04565/2020) and project PRECISE-Accelerating progress toward the new era of precision medicine (PAC-PRECISE-LISBOA-01-0145-FEDER-016394, SAICTPAC/0021/2015). João C. Silva is also grateful to FCT for financial support through the scholarship SFRH/BD/105771/2014.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
This work does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 462 kb)
Rights and permissions
About this article

Cite this article
Silva, J.C., Han, X., Silva, T.P. et al. Glycosaminoglycan remodeling during chondrogenic differentiation of human bone marrow−/synovial-derived mesenchymal stem/stromal cells under normoxia and hypoxia. Glycoconj J 37, 345–360 (2020). https://doi.org/10.1007/s10719-020-09911-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10719-020-09911-5