
Abstract
Synthesis of the pentasaccharide with a 2-aminoethyl linker attached to the reducing end corresponding to the cell wall O-antigen of Escherichia coli O86 strain is reported. The synthetic strategy involves sequential glycosylation of suitably protected monosaccharide intermediates under similar glycosylation reaction conditions. Thioglycosides have been used as glycosyl donor throughout the synthetic strategy. Conformational analysis of the synthesized pentasaccharide has been carried out using 2D ROESY NMR spectral analysis and all atom explicit molecular dynamics (MD) simulation technique.

Facile synthesis of the pentasaccharide with a 2-aminoethyl linker attached to the reducing end corresponding to the cell wall O-antigen of Escherichia coli O86 strain is reported. Conformational analysis of the synthesized pentasaccharide has been carried out using 2D ROESY NMR spectral analysis and all atom explicit molecular dynamics (MD) simulation technique.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Development of novel therapeutics against bacterial infections is the thrust area of medicinal chemistry and drug discovery program. Emergence of bacterial strains resistant to the commonly used antibiotics poses extra challenges to this [1]. As an alternative approach for the eradication of bacterial infections several glycoconjugate based promising vaccine candidates have been developed in the recent past [2–5], in which bacterial capsular polysaccharides (CPSs) and lipopolysaccharides (LPSs) have been used as the major component. CPS and LPS are important constituents of the bacterial cell wall [6] and play important roles during the initial stage of bacterial infections to the host [7]. In Gram-negative bacteria the cell wall LPS is composed of lipid A, core oligosaccharide and O-antigens [8]. The O-antigens, which are exposed outer layer of the cell wall, are mostly responsible for the pathogenicity of the bacteria. The O-antigens are composed of oligosaccharide repeating units, which consist of a variety of monosaccharides. Escherichia coli (E. coli) O86 is an enteropathogenic strain, which causes enteric or diarrheal infections in human [9]. Till date, two different serotypes of this strain have been reported such as E. coli O86:K2:H2 [10] and E. coli O86:K62:B7 [11] having similar O-polysaccharide repeating units consisting of D-galactose, D-galactosamine and L-fucose with appropriate stereochemistry at the glycosyl linkages. The structures of the repeating units of the O-antigens of E. coli O86 are similar to the human blood group antigens and the immunochemical studies showed that both strains produce anti-B antibodies [11]. In order to get a better understanding of the antigenicity of the glycoconjugate of the O-antigen of E. coli O86:K62:B7, it is essential to have significant quantity of the pure oligosaccharide hapten for its use in a variety of immunological experiments. Isolation of the oligosaccharide from the killed bacterial cell wall is tedious and suffers from several shortcomings such as handling of live bacterial strain, difficult to remove biological impurities, batch to batch variation in the isolated oligosaccharide chain etc. Therefore, development of a concise chemical synthetic strategy would be the best option to get structurally pure oligosaccharide repeating unit corresponding to the E. coli O86:K62:B7 for its immunological studies and further use in the glycoconjugate preparation. The target pentasaccharide repeating unit contains three 1,2-cis glycosyl linkages, which makes the synthesis quite challenging due to the problems associated with the formation of 1,2-cis glycosidic bonds [12]. Very recently, Tony Mong et al. [13] reported the synthesis of this pentasaccharide repeating unit as its methyl glycoside applying specially designed reaction conditions for the glycosylations. However, the methoxy group present at the reducing end of the synthesized pentasaccharide cannot be easily removed keeping the cyclic structure of the sugar moiety intact at the reducing end for its further modification towards the preparation of a glycoconjugate derivative. Therefore, it was decided to synthesize the pentasaccharide repeating unit using minimum number of steps and generalized reaction conditions keeping a 2-aminoethyl group at the reducing end, which provides ready availability of an amino group for the conjugation with a suitable protein using standard reaction conditions. Understanding the structural insights of a molecule at an atomic resolution is vital to correlate its biological function. In order to get information on the conformational behaviour of the synthesized pentasaccharide in aqueous environment, NOE based NMR spectral analysis in conjugation with molecular dynamics simulation (MD) techniques have been carried out. Concise chemical synthesis of the pentasaccharide repeating unit as its 2-aminoethyl glycoside corresponding to the O-antigen of E. coli O86:K62:B7 and its conformational studies are presented herein.
Structure of the repeating unit of the O-specific polysaccharide of E. coli O86:K62:B7 [11].
Results and discussion
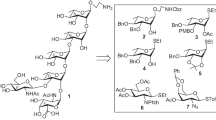
The target pentasaccharide 1 as its 2-aminoethyl glycoside was synthesized using a linear glycosylation strategy. A number of suitably functionalized monosaccharide derivatives 3 [14], 4 [15], 5, 6 [16] and 7 [17] were prepared from the commercially available reducing sugars using recently reported reaction conditions (Fig. 1). A number of recently reported reaction methodologies have been applied in the synthetic strategy. The key feathers of this report include (a) sequential linear glycosylations; (b) use of thioglycoside donor; (c) use of a combination of N-iodosuccinimide (NIS) and a catalytic amount of perchloric acid supported over silica gel (HClO4-SiO2) as thiophilic activator; (d) use of HClO4-SiO2 as a noncorrosive solid acid catalyst; (e) removal of benzylidene acetal and acetylation in one step; (f) use of a 2-aminoethyl linker at the reducing terminal and (g) conformational analysis using NMR spectroscopy and molecular dynamics simulation studies.

Structure of the synthesized pentasaccharide as its 2-aminoethyl glycoside and its synthetic intermediates
Ethyl 2-O-acetyl-3-O-allyl-4,6-O-benzylidene-1-thio-β-D-galactopyranoside (5) was prepared in 98 % yield by the acetylation of ethyl 3-O-allyl-4,6-O-benzylidene-1-thio-β-D-galactopyranoside (2) [18] using acetic anhydride and pyridine (Scheme 1).

Reagents: acetic anhydride, pyridine, room temperature, 2 h, 98 %
Stereoselective 1,2-cis glycosylation of D-galactosamine derivative (3) with D-galactosamine thioglycoside derivative (4) in the presence of a combination [19] of N-iodosuccinimide (NIS) and HClO4-SiO2 [20] furnished disaccharide derivative 8 in 72 % yield, which was confirmed from its spectral analysis [signals at δ 5.40 (d, J = 3.0 Hz, H-1B), 4.46 (d, J = 8.0 Hz, H-1A) in the 1H NMR and δ 100.6 (C-1A), 94.8 (C-1B) in the 13C NMR spectra]. Presence of a non-participating azido group at C-2 position of the glycosyl donor (4) supported the formation of 1,2-cis glycosylated product. De-O-acetylation of compound 8 using sodium methoxide [21] afforded disaccharide acceptor 9 in 93 % yield. 1,2-Trans selective glycosylation of compound 9 with D-galactose thioglycoside derivative 5 in the presence of a combination [19] of NIS and HClO4-SiO2 [20] at low temperature resulted in the formation of trisaccharide derivative 10 in 70 % yield. Formation of compound 10 was supported from its spectral analysis [signals at δ 5.33 (d, J = 3.0 Hz, H-1B), 4.87 (d, J = 7.5 Hz, H-1C), 4.33 (d, J = 8.0 Hz, H-1A) in the 1H NMR and δ 102.1 (C-1A), 101.9 (C-1C), 94.5 (C-1B) in the 13C NMR spectra]. Presence of O-acetyl group at C-2 position of the donor (5) having neighbouring group participation ability directed the formation of 1,2-trans glycoside. De-O-acetylation of the acetyl group in compound 10 using sodium methoxide [21] gave trisaccharide acceptor 11 in 87 % yield. Stereoselective 1,2-cis glycosylation of compound 11 with L-fucose thioglycoside derivative 6 in the presence of a combination [19] of NIS and HClO4-SiO2 in a mixed solvent (diethyl ether-dichloromethane) led to the formation of the required tetrasaccharide derivative together with its other isomeric product in minor quantity (~10 %). The crude product mixture was treated with acetic anhydride in the presence of HClO4-SiO2 to convert the benzylidene acetals into O-acetate groups in one step [22]. Purification of the acetylated products furnished tetrasaccharide 12 in 70 % overall yield. Spectral analysis of the compound 12 unambiguously confirmed its formation [signals at δ 5.45 (d, J = 3.5 Hz, H-1D), 5.21 (d, J = 3.0 Hz, H-1B), 4.62 (d, J = 7.5 Hz, H-1C), 4.23 (d, J = 8.5 Hz, H-1A) in the 1HNMR and δ 102.5 (C-1C), 102.4 (C-1A), 97.6 (C-1D), 94.2 (C-1B) in the 13CNMR spectra]. The formation of 1,2-cis glycosylated product was obtained by keeping a non-participating benzyl ether at the C-2 position of the glycosyl donor (6) as well as using ether in the reaction solvent [23, 24]. Removal of the allyl group from compound 12 by the treatment with palladium chloride [25] afforded tetrasaccharide acceptor 13 in 76 % yield. Further 1,2-cis glycosylation of compound 13 with D-galactose thioglycoside 7 in the presence of a combination [19] of NIS and HClO4-SiO2 [20] in a mixed solvent (diethyl ether-dichloromethane) led to the formation of pentasaccharide derivative 14 in 71 % yield. Spectroscopic studies of compound 14 supported its formation [signals at δ 5.36 (d, J = 3.5 Hz, H-1B), 5.34 (d, J = 3.0 Hz, H-1D), 5.29 (d, J = 3.5 Hz, H-1E), 4.58 (d, J = 7.5 Hz, H-1C), 4.25 (d, J = 7.5 Hz, H-1A) in the 1HNMR and δ 102.7 (C-1A), 102.3 (C-1C), 97.8 (C-1B), 94.4 (2 C, C-1D, C-1E) in the 13CNMR spectra]. The formation of 1,2-cis glycosylated product was obtained by keeping a non-participating benzyl ether at the C-2 position of the glycosyl donor (6) as well as tuning the reaction condition using ether in the reaction solvent [23, 24]. Finally, compound 14 was subjected to a sequence of functional group transformations, which include (a) direct transformation of azido group into acetamido group by treatment with thioacetic acid [26]; (b) de-O-acetylation using sodium methoxide and (c) removal of benzyl ether, benzylidene acetal and Cbz group under a catalytic transfer hydrogenation [27] using triethylsilane and 20 % palladium hydroxide on charcoal to furnish the deprotected target pentasaccharide 1 in 50 % over all yield. NMR spectral analysis of compound 1 supported its formation [signals at δ 5.26 (d, J = 3.5 Hz, H-1D), 5.25 (d, J = 3.5 Hz, H-1E), 5.06 (d, J = 3.5 Hz, H-1B), 4.72 (d, J = 8.0 Hz, H-1C), 4.60 (d, J = 8.0 Hz, H-1A) in the 1HNMR and δ 102.2 (C-1C), 100.9 (C-1A), 98.8 (C-1D), 92.9 (2 C, C-1B, C-1E) in the 13C NMR spectra] (Scheme 2).

Reagents: a NIS, HClO4-SiO2, MS 4 Å, CH2Cl2, − 10 °C, 1 h, 72 %; b 0.1 M CH3ONa, CH3OH, room temperature, 2 h, 93 % for compound 9 and 87 % for compound 11; c NIS, HClO4-SiO2, MS 4 Å, CH2Cl2, − 25 °C, 1 h, 70 %; d NIS, HClO4-SiO2, Et2O-CH2Cl2 (2:1), − 5 °C, 1 h; e acetic anhydride, HClO4-SiO2, room temperature, 20 min, 70 % in two steps; f PdCl2, CH3OH, 0–5 °C, 1.5 h, 76 %; g NIS, HClO4-SiO2, Et2O-CH2Cl2 (3:1), − 8 °C, 1 h, 71 %; h CH3COSH, pyridine, room temperature, 22 h; i 0.1 M CH3ONa, CH3OH, room temperature, 2 h; (j) 20 % Pd(OH)2-C, Et3SiH, CH3OH, room temperature, 24 h, overall 50 %
Conformational analysis
In the present study, NMR spectroscopy in conjunction with the Molecular Dynamics (MD) simulation was used to determine the three-dimensional conformational analysis of carbohydrate structure [28]. The distance information of the molecule, which is generally used to determine the three-dimensional structure of any molecule, was collected from two-dimensional 1H-1H ROESY experiments with 150 ms mixing time. This information was further used in MD simulation to determine the three-dimensional structure of the molecule in solution at an atomic resolution. The computational sampling of conformation was performed for a time scale of 20 ns with inter- as well as intra-glycosidic ROE informations. A schematic presentation of the inter- and intra-glycosidic NOE in compound 1 is presented in Fig. S1 in supporting information. The proton-proton distances observed from the MD simulation (Fig. 2a, b) at the interglycosidic linkages were in good agreement to the ROEs cross peaks observed in the 2D ROESY spectrum. In particular, the inter-glycosidic linkages obtained for H-4A/H-1B, H-3B/H-1C, H-2C/H-1D, and H-4C/H-1E appeared to be within the range of 2.3 Å – 3.0 Å distance. These informations, which were conserved in the course of MD simulation, were also reflective to the fact that compound 1 attained rigid conformation to the glycosidic linkages between β-D-GalpNAc (ring A) and α-D-GalpNAc (ring B); as well as between α-D-GalpNAc (ring B) and β-D-Galp (ring C). Correspondingly strong ROEs were also observed in the NMR spectrum that reflected efficient spin-spin relaxation effect. The distances for the interglycosidic linkages of H-2A/H-1B, H-6A/H-1B, and H-6C/H-1E were found to be greater than 4 Å, which conferred higher degree of freedom (conformational flexibility). In addition, larger separation distance persisted between β-D-GalpNAc (ring A) and α-D-GalpNAc (ring B) as well as between β-D-Galp (ring C) and α-D-Galp (ring E). Notably, the distance between glycosidic proton H-1A and proton of protecting group (NHAc) (Fig. 2a) is found to be within 2.7 Å – 3.5 Å that confirmed a rigid conformation around the linkage. The molecular conformational sampling was also evaluated with the consideration of torsion angle (dihedral angle of oligosaccharides) using the notion Phi angle (φn) as (Hn-Cn-On-Cn+1) and Psi angle (Ψn) as (Cn-On-Cn+1-Hn+1), where n refers to the ring number, considered for the calculation. The obtained torsion angles were within the range of −100° to +10° between β-D-GalpNAc (ring A) and α-D-GalpNAc (ring B) as well as between β-D-Galp (ring C) and α-D-Galp (ring E). Likewise, the torsion angles appeared to be within the range of +10° to +100° between B-C and C-D rings (Fig. 2c, d). Moreover, the confined density of the scatter plot obtained for the φ and Ψ correlation also indicated that these glycosidic linkages were within a rigid three-dimensional space (Fig. 2c, d). The overall root-mean-squared-deviation (RMSD) of compound 1 as indicated from the overall trajectory analysis was reflective to be in a range of 0.35 Å to 1.8 Å. It is noteworthy that the conformational deviation (RMSD) within a scale of 0–4 Å is considered to be almost identical for all the (bio) organic molecules with respect to its structural dynamicity and molecular function [29]. The superimposed snapshots of compound 1 are also displayed in Fig. 3, which dictates the stability of conformation. More importantly, the conformational rigidity is reflective for all the rings except for α-D-Galp (ring E).

Account of distance information for interglycosidic linkages a and b, and sugar ring dihedral angles (Phi and Psi) of compound 1 c and d

Representation of conformational snapshots of compound 1 obtained using MD simulation
Conclusion
In summary, the pentasaccharide repeating unit corresponding to the cell wall O-antigen of E. coli O86:K62:B7 has been synthesized in good yield using linear glycosylation sequence. Compound 1 with a 2-aminoethyl linker at the reducing terminal has been synthesized using minimum number of reaction steps with very good yield. The conformational analysis of the pentasaccharide was carried out using MD simulation technique in conjugation with 2D ROESY NMR spectral analysis. The conformational analysis confirmed that the compound 1 is significantly rigid in solution.
Experimental section
General methods
All reactions were monitored by thin layer chromatography over silica gel coated TLC plates. The spots on TLC were visualized by warming ceric sulphate (2 % Ce(SO4)2 in 2 N H2SO4) sprayed plates in hot plate. Silica gel 230–400 mesh was used for column chromatography. NMR spectra were recorded on Bruker Avance 500 MHz using CDCl3 as solvent and TMS as internal reference unless stated otherwise. Chemical shift value is expressed in δ ppm. The complete assignment of proton and carbon spectra was carried out by using a standard set of NMR experiments, e.g. 1H NMR, 13C NMR, 13C DEPT 135, 2D COSY and 2D HSQC etc. In addition, 2D ROESY (300 ms mixing time) was performed to assist in the conformational analysis. The ROESY experiments were performed with 456 increments in t1 and 2 K data points in t2. The spectral width was normally 10 ppm in both dimensions. After 16 dummy scans, 80 scans were recorded per t1 increment. After zero-filling in t1, 4 K (t2) × 1 K (t1) data matrices were obtained. The two-dimensional NMR data were processed by Top Spin software suite (Bruker, Switzerland). MALDI-MS were recorded on a Bruker Daltronics mass spectrometer. Optical rotations were recorded in a Jasco P-2000 spectrometer. Commercially available grades of organic solvents of adequate purity are used in all reactions. HClO4-SiO2 was prepared using the report of Chakraborti et al. [20].
Preparation of HClO 4 -SiO 2 [20]
HClO4 (1.8 g, 12.5 mmol, as a 70 % aq solution) was added to a suspension of SiO2 (230–400 mesh, 23.7 g) in Et2O (70.0 mL). The mixture was concentrated and the residue was heated at 100 °C for 72 h under vacuum to furnish HClO4-SiO2 (0.5 mmol/g) as a free flowing powder. In all glycosylation reactions HClO4-SiO2 has been used in catalytic quantity (e.g. 3–5 mg HClO4-SiO2 per 100 mg of NIS).
Ethyl 2- O -acetyl-3- O -allyl-4,6- O -benzylidene-1-thio-β-D-galactopyranoside (5)
A solution of compound 2 (3.0 g, 8.51 mmol) in acetic anhydride (5 mL) and pyridine (5 mL) was allowed to stir at room temperature for 2 h. The solvents were removed under reduced pressure and the crude product was passed through a short pad of SiO2 using hexane-EtOAc (2:1) as eluant to give pure compound 5 (3.3 g, 98 %). White solid; m.p. 124–125 °C [EtOH]; [α]D 25 + 0.5 (c 1.0, CHCl3); IR (KBr): 3026, 2902, 1620, 1515, 1466, 1230, 757, 697 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.61–7.44 (m, 5H, Ar-H), 6.08–5.92 (m, 1 H, CH = CH2), 5.62 (s, 1 H, PhCH), 5.51 (t, J = 10.0 Hz, 1 H, H-2), 5.40–5.27 (m, 2 H, CH = CH 2), 4.48 (d, J = 9.5 Hz, 1 H, H-1), 4.59 (dd, J = 12.5, 1.5 Hz, 1 H, H-6a), 4.38 (d, J = 3.0 Hz, 1 H, H-4), 4.30–4.18 (m, 2 H, OCH 2CH=), 4.13 (dd, J = 12.5, 2.0 Hz, 1 H, H-6b), 3.67 (dd, J = 9.5, 3.0 Hz, 1 H, H-3), 3.56–3.54 (m, 1 H, H-5), 3.05–2.95 (m, 1 H, SCH 2CH3), 2.86–2.79 (m, 1 H, SCH 2CH3), 2.21 (s, 3 H, COCH 3), 1.40 (t, J = 7.5 Hz, 3 H, SCH2CH 3); 13C NMR (125 MHz, CDCl3): δ 169.2 (COCH3), 137.7–117.2 (Ar-C, CH = CH), 101.3 (PhCH), 82.8 (C-1), 78.4 (C-4), 73.8 (C-3), 70.5 (OCH2CH=), 70.1 (C-5), 69.3 (C-6), 68.0 (C-2), 22.4 (SCH2CH3), 21.0 (COCH3), 14.8 (SCH2 CH3); ESI-MS: 417.1 [M + Na]+; Anal. Calcd. for C20H26O6S (394.48): C, 60.89; H, 6.64 %; found: C, 60.73; H, 6.85 %.
2-(N-Benzyloxycarbonyl)aminoethyl O-(3-O-acetyl-2-azido-4,6-O-benzylidene-2-deoxy-α-D-galactopyranosyl)-(1 → 3)-2-azido-4,6-O-benzylidene-2-deoxy-β-D-galactopyranoside (8)
To a solution of compound 3 (1.5 g, 3.19 mmol) and compound 4 (1.5 g, 3.39 mmol) in anhydrous CH2Cl2 (15 mL) was added MS 4 Å (3 g) and the reaction mixture was cooled to −10 °C. To the cooled reaction mixture were added N-iodosuccinimide (NIS; 800 mg, 3.55 mmol) and HClO4-SiO2 (25 mg) and the reaction mixture was allowed to stir at same temperature for 1 h. The reaction mixture was filtered through a Celite bed and washed with CH2Cl2 (100 mL). The combined organic layer was successively washed with 5 % Na2S2O3 (50 mL), satd. NaHCO3 (50 mL) and H2O (50 mL), dried (Na2SO4) and concentrated under reduced pressure. The crude product was purified over SiO2 using hexane-EtOAc (4:1) as eluant to give pure compound 8 (1.8 g, 72 %). White solid; m.p. 130 °C [EtOH]; [α]D 25 + 12.5 (c 1.0, CHCl3); IR (KBr): 3014, 2932, 1741, 1506, 1457, 1216, 1056, 756, 699 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.37–7.30 (m, 15 H, Ar-H), 5.67 (s, 1 H, PhCH), 5.66 (s, 1 H, PhCH), 5.50 (dd, J = 10.5, 3.0 Hz, 1 H, H-3B), 5.48–5.45 (m, 1 H, NH), 5.40 (d, J = 3.0 Hz, 1 H, H-1B), 5.22 (br s, 2 H, Cbz), 4.67 (d, J = 2.5 Hz, 1 H, H-4B), 4.46 (d, J = 8.0 Hz, 1 H, H-1A), 4.44–4.36 (m, 3 H, H-4A, H-6aA, H-6bA), 4.25–4.16 (m, 3 H, H-5A, H-6bA, H-6bB), 4.15–4.10 (m, 1 H, OCH), 4.07 (dd, J = 10.0, 3.0 Hz, 1 H, H-2B), 4.01 (t, J = 10.5 Hz, 1 H, H-2A), 3.87–3.82 (m, 1 H, OCH), 3.76 (dd, J = 10.0, 3.0 Hz, 1 H, H-3A), 3.65–3.52 (m, 2 H, NCH 2), 3.50–3.48 (m, 1 H, H-5B), 2.14 (s, 3 H, COCH 3); 13C NMR (125 MHz, CDCl3): δ 170.3 (COCH3), 137.5–126.0 (Ar-C), 100.6 (C-1A), 100.9 (PhCH), 100.8 (PhCH), 94.8 (C-1B), 74.0 (C-3A), 73.4 (C-4B), 70.6 (C-4A), 69.5 (OCH2), 69.1 (2 C, C-6A, C-6B), 69.0 (C-3B), 66.7 (Cbz), 66.5 (C-5B), 63.1 (C-5A), 61.3 (C-2A), 56.5 (C-2B), 40.9 (NCH2), 20.9 (COCH3); MALDI-MS: 810.2 [M + Na]+; Anal. Calcd. for C38H41N7O12 (787.77): C, 57.94; H, 5.25 %; found: 57.78; H, 5.43 %.
2-(N-Benzyloxycarbonyl)aminoethyl O-(2-azido-4,6-O-benzylidene-2-deoxy-α-D-galactopyranosyl)-(1 → 3)-2-azido-4,6-O-benzylidene-2-deoxy-β-D-galactopyranoside (9)
A solution of compound 8 (1.7 g, 2.16 mmol) in 0.1 M CH3ONa in CH3OH (40 mL) was stirred at room temperature for 2 h and neutralized with Dowex 50 W X8 (H+) resin. The reaction mixture was filtered through a Celite bed and evaporated to dryness. The crude product was passed through a short pad of SiO2 using hexane-EtOAc (3:1) as eluant to give pure compound 9 (1.5 g, 93 %). White solid; m.p. 124–125 °C [EtOH]; [α]D 25 + 0.5 (c 1.0, CHCl3); IR (KBr): 3018, 2918, 1509, 1457, 1218, 1049, 756, 699 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.54–7.33 (m, 15 H, Ar-H), 5.56 (s, 2 H, 2 PhCH), 5.33 (m, 1 H, NH), 5.20 (s, 1 H, H-1B), 5.10 (br s, 2 H, Cbz), 4.32 (d, J = 8.0 Hz, 1 H, H-1A), 4.30–4.24 (m, 5 H, H-3B, H-4A, H-4B, H-6aA, H-6aB), 4.10–3.99 (m, 3 H, H-5A, H-6bA, H-6bB), 3.98–3.91 (m, 1 H, OCH), 3.85 (t, J = 10.5 Hz, 1 H, H-2A), 3.75–3.69 (m, 1 H, OCH), 3.60 (dd, J = 10.0, 3.0 Hz, 1 H, H-3A), 3.59 (dd, J = 10.0, 3.0 Hz, 1 H, H-2B), 3.50–3.35 (m, 2 H, NCH 2), 3.35–3.30 (m, 1 H, H-5B); 13C NMR (125 MHz, CDCl3): δ 126.3 (COCbz), 129.5–124.0 (Ar-C), 102.5 (C-1A), 101.3 (PhCH), 100.9 (PhCH), 95.1 (C-1B), 75.5 (C-4B), 74.2 (C-3A), 70.6 (C-4A), 69.5 (OCH2), 69.1 (C-6B), 68.9 (C-6A), 66.7 (C-3B), 66.6 (Cbz), 66.5 (C-5B), 63.4 (C-5A), 61.5 (C-2A), 59.9 (C-2B), 40.1 (NCH2); MALDI-MS: 768.2 [M + Na]+; Anal. Calcd. for C36H39N7O11: C, 57.98; H, 5.27 %; found: C, 57.81; H, 5.42 %.
2-(N-Benzyloxycarbonyl)aminoethyl O-(2-O-acetyl-3-O-allyl-4,6-O-benzylidene-β-D-galactopyranosyl)-(1 → 3)-O-(2-azido-4,6-O-benzylidene-2-deoxy-α-D-galactopyranosyl)-(1 → 3)-2-azido-4,6-O-benzylidene-2-deoxy-β-D-galactopyranoside (10)
To a solution of compound 9 (1.4 g, 1.87 mmol) and compound 5 (815 mg, 2.06 mmol) in anhydrous CH2Cl2 (10 mL) was added MS 4 Å (2 g) and the reaction mixture was cooled to −25 °C. To the cooled reaction mixture were added NIS (470 mg, 2.08 mmol) and HClO4-SiO2 (15 mg) and the reaction mixture was allowed to stir at same temperature for 1 h. The reaction mixture was filtered through a Celite bed and washed with CH2Cl2 (100 mL). The combined organic layer was successively washed with 5 % Na2S2O3 (50 mL), satd. NaHCO3 (50 mL) and H2O (50 mL), dried (Na2SO4) and concentrated under reduced pressure. The crude product was purified over SiO2 using hexane-EtOAc (4:1) as eluant to give pure compound 10 (1.4 g, 70 %). White solid; m.p. 154–155 °C [EtOH]; [α]D 25 + 11.5 (c 1.0, CHCl3); IR (KBr): 3021, 2933, 1740, 1506, 1457, 1219, 1055, 756, 697 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.65–7.39 (m, 20 H, Ar-H), 5.91–6.01 (m, 1 H, CH = CH2), 5.69 (s, 1 H, PhCH), 5.65 (s, 1 H, PhCH), 5.63 (s, 1 H, PhCH), 5.50–5.40 (m, 2 H, NH, H-2C), 5.41–5.36 (m, 1 H, CH = CH), 5.33 (d, J = 3.0 Hz, 1 H, H-1B), 5.28 (dd, J = 10.0, 3.0 Hz, 1 H, CH = CH), 5.22 (br s, 2 H, Cbz), 4.87 (d, J = 7.5 Hz, 1 H, H-1C), 4.65 (d, J = 1.5 Hz, 1 H, H-4A), 4.45–4.34 (m, 6 H, H-3A, H-4B, H-4C, H-6aA, H-6abC), 4.33 (d, J = 8.0 Hz, 1 H, H-1A), 4.30–4.21 (m, 1 H, H-6aB), 4.21–4.10 (m, 4 H, H-6bA, H-6bB, OCH 2CH = CH2), 4.09–4.01 (m, 1 H, OCH), 4.01–3.92 (m, 3 H, H-2A, H-2B, H-5A), 3.88–3.79 (m, 1 H, OCH), 3.75 (dd, J = 10.0, 3.0 Hz, 1 H, H-3B), 3.65 (dd, J = 10.0, 3.0 Hz, 1 H, H-3C), 3.61–3.55 (m, 2 H, NCH 2), 3.55–3.45 (m, 1 H, H-5C), 3.41–3.37 (m, 1 H, H-5B), 2.16 (s, 3 H, COCH 3); 13C NMR (125 MHz, CDCl3): δ 169.1 (COCH3), 156.3 (COCbz), 134.7 (CH = CH2), 129.1–126.1 (Ar-C), 117.1 (CH = CH2), 102.1 (C-1A), 101.9 (C-1C), 101.1 (PhCH), 100.7 (PhCH), 100.4 (PhCH), 94.5 (C-1B), 77.4 (C-3C), 75.9 (C-4A), 73.6 (C-3B, C-4B), 73.4 (C-4C), 74.4 (OCH2CH = CH2), 70.2 (C-3A), 70.1 (C-2C), 69.4 (C-6C), 69.1 (C-6A), 69.0 (C-6B), 68.9 (OCH2-), 66.5 (C-5C, C-5B), 63.9 (C-5A), 61.6 (C-2B), 58.4 (C-2A), 40.9 (NCH2), 20.9 (COCH3); MALDI-MS: 1100.4 [M + Na]+; Anal. Calcd. for C54H59N7O17 (1078.08): C, 60.16; H, 5.52 %; found: C, 60.00; H, 5.70 %.
2-(N-Benzyloxycarbonyl)aminoethyl O-(3-O-allyl-4,6-O-benzylidene-β-D-galactopyranosyl)-(1 → 3)-O-(2-azido-4,6-O-benzylidene-2-deoxy-α-D-galactopyranosyl)-(1 → 3)-2-azido-4,6-O-benzylidene-2-deoxy-β-D-galactopyranoside (11)
A solution of compound 10 (1.2 g, 1.11 mmol) in 0.1 M CH3ONa in CH3OH (30 mL) was stirred at room temperature for 2 h and neutralized with Dowex 50 W X8 (H+) resin. The reaction mixture was filtered through a Celite bed and evaporated to dryness. The crude product was passed through a SiO2 using hexane-EtOAc (3:1) as eluant to give pure compound 11 (1.0 g, 87 %). White solid; m.p. 134–135 °C [EtOH]; [α]D 25 + 11.5 (c 1.0, CHCl3); IR (KBr): 3016, 2917, 1508, 1455, 1216, 1048, 756, 699 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.59–7.21 (m, 20 H, Ar-H), 5.97–5.91 (m, 1 H, CH = CH2), 5.57 (s, 1 H, PhCH), 5.55 (s, 1 H, PhCH), 5.51 (s, 1 H, PhCH), 5.40–5.30 (m, 2 H, NH, CH = CH 2), 5.27 (d, J = 3.0 Hz, 1 H, H-1B), 5.18 (dd, J = 10.5 Hz, 1 H, CH = CH), 5.08 (br s, 2 H, Cbz), 4.57 (d, J = 7.5 Hz, 1 H, H-1C), 4.52 (d, J = 2.5 Hz, 1 H, H-4A), 4.35–4.29 (m, 1 H, H-4B), 4.28 (t, J = 9.5 Hz, 1 H, H-1A), 4.26–4.15 (m, 7 H, H-4C, H-6aA, H-6aB, H-6abC, OCH 2CH = CH2), 4.08–3.91 (m, 6 H, H-2B, H-2C, H-3A, H-6bA, H-6bB, OCH), 3.91–3.95 (m, 1 H, H-5A), 3.85 (t, J = 10.0 Hz, 1 H, H-2A), 3.75–3.66 (m, 1 H, OCH), 3.66 (dd, J = 10.0, 3.0 Hz, 1 H, H-3B), 3.46–3.44 (m, 3 H, H-3C, NCH 2), 3.39–3.35 (m, 1 H, H-5C), 3.31–3.25 (m, 1 H, H-5B); 13C NMR (125 MHz, CDCl3): δ 156.3 (COCbz), 135.0 (CH = CH2) 128.9–126.1 (Ar-C), 117.4 (CH = CH2), 104.2 (C-1C), 102.1 (C-1A), 101.0 (PhCH), 100.8 (2 PhCH), 94.4 (C-1B), 78.4 (C-3C), 76.1 (C-4A), 73.7 (C-3B), 73.6 (C-4B), 73.5 (C-4C), 71.1 (CH2CH = CH2), 70.2 (2 C, C-2C, C-3A), 69.4 (C-6C), 69.2 (C-6A), 68.9 (2 C, C-6B, OCH2), 66.7 (C-5C), 66.6 (C-Cbz), 66.5 (C-5B), 63.9 (C-5A), 61.5 (C-2B), 58.4 (C-2A), 40.9 (NCH2); MALDI-MS: 1058.3 [M + Na]+; Anal. Calcd. for C52H57N7O16 (1036.05): C, 60.28; H, 5.55 %; found: C, 60.11; H, 5.70 %.
2-(N-Benzyloxycarbonyl)aminoethyl O-(2,3,4-tri-O-benzyl-α-L-fucopyranosyl)-(1 → 2)-O-(4,6-di-O-acetyl-3-O-allyl-β-D-galactopyranosyl)-(1 → 3)-O-(4,6-di-O-acetyl-2-azido-2-deoxy-α-D-galactopyranosyl)-(1 → 3)-4,6-di-O-acetyl-2-azido-2-deoxy-β-D-galactopyranoside (12)
To a solution of compound 11 (900 mg, 0.87 mmol) and compound 6 (450 mg, 0.94 mmol) in anhydrous Et2O-CH2Cl2 (10 mL; 2:1) was added MS 4 Å (2 g) and the reaction mixture was cooled to −5 °C. To the cooled reaction mixture were added NIS (220 mg, 0.97 mmol) and HClO4-SiO2 (10 mg) and the reaction mixture was allowed to stir at same temperature for 1 h. The reaction mixture was filtered through a Celite bed and washed with CH2Cl2 (50 mL). The combined organic layer was successively washed with 5 % Na2S2O3 (50 mL), satd. NaHCO3 (50 mL) and H2O (50 mL), dried (Na2SO4) and concentrated under reduced pressure. To a solution of the crude product in acetic anhydride (10 mL) was added HClO4-SiO2 (250 mg) and the reaction mixture was stirred at room temperature for 20 min. The reaction mixture was filtered and concentrated under reduced pressure. The crude product was purified over SiO2 using hexane-EtOAc (4:1) as eluant to give pure compound 12 (880 mg, 70 %). Colorless oil; [α]D 25 + 3.3 (c 1.0, CHCl3); IR (KBr): 3456, 2922, 2875, 2111, 1735, 1719, 1510, 1456, 1392, 1275, 1232, 1090, 1070, 1028, 709 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.34–7.22 (m, 20 H, Ar-H), 5.79–5.69 (m, 1 H, CH = CH2), 5.68 (d, J = 2.5 Hz, 1 H, H-4C), 5.45 (d, J = 3.5 Hz, 1 H, H-1D), 5.41 (d, J = 2.0 Hz, 1 H, H-4A), 5.33 (d, J = 3.5 Hz, 1 H, H-4B), 5.30–5.15 (m, 1 H, NH), 5.21 (d, J = 3.0 Hz, 1 H, H-1B), 5.18–5.11 (m, 1 H, CH = CH), 5.10 (br s, 2 H, Cbz), 5.08 (dd, J = 10.5, 3.0 Hz, 1 H, CH = CH), 4.90 (d, J = 11.5 Hz, 1 H, PhCH), 4.79–4.65 (4 d, J = 11.5 Hz, 4 H, 2 PhCH), 4.62 (d, J = 7.5 Hz, 1 H, H-1C), 4.57 (d, J = 11.5 Hz, 1 H, PhCH) 4.39–4.30 (m, 2 H, H-3D, H-5D), 4.23 (d, J = 8.5 Hz, 1 H, H-1A), 4.19–4.18 (m, 1 H, H-6aA), 4.20–4.05 (m, 7 H, H-6bA, H-6abC, H-6abB, CH 2CH = CH2), 4.05–3.99 (2 dd, J = 10.0, 3.0 Hz, 2 H, H-2D, H-3B), 3.99–3.90 (m, 1 H, OCH), 3.85–3.70 (m, 5 H, H-2C, H-3A, H-3C, H-5A, H-5C), 3.70–3.47 (m, 4 H, H-2A, H-4D, H-5B, OCH), 3.46–3.42 (m, 2 H, NCH 2), 3.41 (dd, J = 10.5, 3.0 Hz, 1 H, H-2B), 2.14, 2.10, 2.08, 2.03 (4 s, 18 H, 6 COCH 3), 1.19 (d, J = 6.5 Hz, 3 H, CCH 3); 13C NMR (125 MHz, CDCl3): δ 170.0 (6 COCH3), 156.3 (COCbz), 134.1 (CH = CH2), 128.5–127.3 (Ar-C), 116.1 (CH = CH2), 102.5 (C-1C), 102.4 (C-1A), 97.6 (C-1D), 94.2 (C-1B), 79.9 (C-3C), 79.5 (C-3B), 77.6 (C-4D), 75.6 (C-3D), 74.7 (PhCH2), 74.3 (PhCH2), 72.9 (C-5D), 72.8 (C-2D), 72.6 (2C, OCH2, PhCH2), 70.8 (C-3A), 70.7 (C-2C), 70.5 (CH2CH = CH2), 69.9 (C-4C), 69.7 (C-5C), 68.1 (C-5B), 66.8 (Cbz), 66.1 (C-4B), 65.4 (C-4A), 63.4 (C-5A), 62.5 (C-6C), 62.1 (C-6B), 61.9 (C-6A), 61.3 (C-2A), 58.9 (C-2B), 40.9 (NCH2), 20.8, 20.7, 20.5 (6 COCH3), 16.7 (CCH3); MALDI-MS: 1462.5 [M + Na]+; Anal. Calcd. for C70H85N7O26 (1440.46): C, 58.37; H, 5.95 %; found: C, 58.20; H, 6.15 %.
2-(N-Benzyloxycarbonyl)aminoethyl O-(2,3,4-tri-O-benzyl-α-L-fucopyranosyl)-(1 → 2)-O-(4,6-di-O-acetyl-β-D-galactopyranosyl)-(1 → 3)-O-(4,6-di-O-acetyl-2-azido-2-deoxy-α-D-galactopyranosyl)-(1 → 3)-4,6-di-O-acetyl-2-azido-2-deoxy-β-D-galactopyranoside (13)
To a solution of compound 12 (800 mg, 0.56 mmol) in dry CH3OH (10 mL) was added PdCl2 (50 mg, 0.28 mmol) and the reaction mixture was allowed to stir at 0–5 °C for 1.5 h. The solvents were removed under reduced pressure and the crude product was purified over SiO2 using hexane-EtOAc (3:1) as eluant to give pure compound 13 (600 mg, 76 %). Colorless oil; [α]D 25 + 3.4 (c 1.0, CHCl3); IR (KBr): 3434, 2112, 1741, 1275, 1247, 1132, 1069, 1029, 755, 709 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.44–7.34 (m, 20 H, Ar-H), 5.56 (d, J = 2.5 Hz, 1 H, H-4C), 5.49 (s, 1 H, H-4A), 5.41 (d, J = 3.0 Hz, 1 H, H-4B), 5.39–5.31 (m, 1 H, NH), 5.30 (d, J = 3.5 Hz, 1 H, H-1B), 5.20 (br s, 2 H, Cbz), 5.02 (d, J = 12.0 Hz, 1 H, PhCH), 4.99 (d, J = 12.0 Hz, 1 H, PhCH), 4.97 (d, J = 3.0 Hz, 1 H, H-1D), 4.88–4.78 (m, 3 H, 3 PhCH), 4.71 (d, J = 12.0 Hz, 1 H, PhCH), 4.62 (d, J = 7.5 Hz, 1 H, H-1C), 4.42–4.38 (m, 1 H, H-5D), 4.38–4.32 (m, 1 H, H-1A), 4.31–4.20 (m, 5 H, H-3D, H-4D, H-6aA, H-6abC), 4.19–4.09 (m, 6 H, H-2D, H-3C, H-5A, H-6abB, H-6bA), 4.08–3.98 (m, 1 H, OCH), 3.92–3.80 (m, 4 H, H-3A, H-3B, H-5C, OCH), 3.78–3.70 (m, 2 H, H-2A, H-5B), 3.68 (dd, J = 11.0, 3.5 Hz, H-2B), 3.60–3.48 (m, 3 H, H-2C, NCH 2), 2.29, 2.24, 2.22, 2.18, 2.17 (5 s, 18 H, 6 COCH 3), 1.23 (d, J = 6.5 Hz, 3 H, CCH 3); 13C NMR (125 MHz, CDCl3): δ 170.1–169.3 (6 COCH3), 156.1 (COCbz), 128.4–127.4 (Ar-C), 102.4 (C-1A), 101.2 (C-1C), 101.1 (C-1B), 93.7 (C-1D), 81.1 (C-2C), 79.8 (C-3C), 77.5 (C-2A), 76.5 (C-4D), 74.7 (PhCH2), 74.4 (PhCH2) 72.6 (PhCH2), 72.5 (C-2D), 72.1 (C-3B), 71.4 (C-3D), 71.1 (C-3A), 70.9 (C-5C), 69.8 (OCH2), 69.5 (C-4C), 68.5 (C-4B), 68.1 (C-5D), 67.1 (C-5A), 66.6 (Cbz), 63.2 (C-4A), 62.3 (C-6B), 62.1 (C-5B), 61.3 (C-6C), 61.2 (C-6A), 59.5 (C-2B), 40.9 (NCH2), 20.7, 20.5, 20.4, 20.2 (6 COCH3), 16.9 (CCH3); MALDI-MS: 1422.5 [M + Na]+; Anal. Calcd. for C67H81N7O26 (1400.39): C, 57.46; H, 5.83 %; found: C, 57.30; H, 6.05 %.
2-(N-Benzyloxycarbonyl)aminoethyl O-(2,3-di-O-benzyl-4,6-O-benzylidene-α-D-galactopyranosyl)-(1 → 3)-O-[(2,3,4-tri-O-benzyl-α-L-fucopyranosyl)-(1 → 2)-O]-(4,6-di-O-acetyl-β-D-galactopyranosyl)-(1 → 3)-O-(4,6-di-O-acetyl-2-azido-2-deoxy-α-D-galactopyranosyl)-(1 → 3)-4,6-di-O-acetyl-2-azido-2-deoxy-β-D-galactopyranoside (14)
To a solution of compound 13 (500 mg, 0.36 mmol) and compound 7 (190 mg, 0.38 mmol) in anhydrous Et2O-CH2Cl2 (6 mL; 3:1) was added MS 4 Å (1 g) and the reaction mixture was cooled to −8 °C. To the cooled reaction mixture were added NIS (90 mg, 0.40 mmol) and HClO4-SiO2 (5 mg) and the reaction mixture was allowed to stir at same temperature for 1 h. The reaction mixture was filtered through a Celite bed and washed with CH2Cl2 (25 mL). The combined organic layer was successively washed with 5 % Na2S2O3 (25 mL), satd. NaHCO3 (25 mL) and H2O (25 mL), dried (Na2SO4) and concentrated under reduced pressure. The crude product was purified over SiO2 using hexane-EtOAc (3:1) as eluant to give pure compound 14 (470 mg, 71 %). Colorless oil; [α]D 25 + 4.9 (c 1.0, CHCl3); IR (KBr): 3437, 2110, 1741, 1274, 1250, 1130, 1073, 1027, 756, 709 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.44–7.23 (m, 35 H, Ar-H), 5.61 (d, J = 2.5 Hz, 1 H, H-4B), 5.42–5.19 (m, 2 H, H-4A, H-4C), 5.36 (d, J = 3.5 Hz, 1 H, H-1B), 5.34 (d, J = 3.0 Hz, 1 H, H-1D), 5.29 (d, J = 3.5 Hz, 1 H, H-1E), 5.24–5.20 (m, 1 H, NH), 5.08 (br s, 2 H, Cbz), 5.00 (s, 1 H, PhCH), 4.98–4.60 (9 d, J = 12.0 Hz, 9 H, 9 PhCH), 4.58 (d, J = 7.5 Hz, 1 H, H-1C), 4.58–4.56 (m, 1 H, H-4E), 4.50–4.41 (m, 1 H, H-5D), 4.39 (d, J = 11.5 Hz, 1 H, PhCH), 4.35–4.24 (m, 1 H, H-4D), 4.25 (d, J = 7.5 Hz, 1 H, H-1A), 4.21–3.99 (m, 10 H, H-3C, H-3D, H-3E, H-6abA, H-6abB, H-6abE, OCH), 3.99–3.70 (m, 10 H, H-2A, H-2B, H-2D, H-3A, H-3B, H-5A, H-5B, H-6abC, OCH), 3.70–3.63 (m, 1 H, H-5C), 3.58–3.40 (m, 2 H, H-2C, H-5E), 3.39 (dd, J = 10.0, 3.0 Hz, 1 H, H-2E), 2.64, 2.14, 2.07, 2.06, 2.05 (5 s, 18 H, 6 COCH 3), 1.22 (d, J = 6.5 Hz, 3 H, CCH 3); 13C NMR (125 MHz, CDCl3): δ 170.1 (5 C, 5 COCH3), 168.7 (COCH3), 156.3 (COCbz), 128.7–126.3 (Ar-C), 102.7 (C-1A), 102.3 (C-1C), 100.5 (PhCH), 97.8 (C-1B), 94.4 (2 C, C-1D, C-1E), 80.7 (C-3C), 77.9 (C-2A), 77.1 (C-4D), 76.1 (C-3E), 75.5 (C-2D), 75.4 (C-3B), 74.9 (PhCH2), 74.5 (PhCH2), 73.5 (PhCH2), 72.9 (C-3A), 72.6 (C-3D), 72.5 (PhCH2), 72.4 (PhCH2), 71.8 (C-5C), 70.9 (C-5A), 70.8 (C-2C), 70.5 (C-4E), 69.9 (OCH2), 68.7 (C-6E), 68.2 (C-4B), 66.8 (Cbz), 66.2 (C-5D), 65.7 (C-4C), 63.5 (C-4A), 63.1 (C-5B), 62.4 (C-6B), 62.2 (C-5E), 61.6 (C-6C), 61.3 (C-6A), 58.4 (C-2E), 40.9 (NCH2), 20.7 (6 COCH3), 16.7 (CCH3); MALDI-MS: 1852.7 [M + Na]+; Anal. Calcd. for C94H107N7O31 (1830.88): C, 61.66; H, 5.89 %; found: C, 61.50; H, 6.10 %.
2-aminoethyl O-(α-D-galactopyranosyl)-(1 → 3)-O-[(α-L-fucopyranosyl)-(1 → 2)-O]-(β-D-galactopyranosyl)-(1 → 3)-O-(2-acetamido-2-deoxy-α-D-galactopyranosyl)-(1 → 3)-2-acetamiso-2-deoxy-β-D-galactopyranoside (1)
To a solution of compound 14 (400 mg, 0.22 mmol) in pyridine (0.5 mL) was added CH3COSH (0.2 mL, 2.78 mmol) and the reaction mixture was stirred at room temperature for 22 h. The reaction mixture was evaporated under reduced pressure and co-evaporated with toluene (3x50 mL). A solution of the crude product in 0.1 M CH3ONa in CH3OH (10 mL) was stirred at room temperature for 2 h, neutralized with Dowex 50 W-X8 (H+) resin, filtered and concentrated to dryness. The crude product was passed through a short pad of SiO2 using EtOAc as eluant to give de-O-acetylated product. To a solution of the product in CH3OH (5 mL) were added 20 % Pd(OH)2-C (100 mg) and Et3SiH (2 mL, 12.52 mmol) and the reaction mixture was allowed to stir at room temperature for 24 h. The reaction mixture was filtered through a Celite bed and the filtering bed was washed with CH3OH-H2O (20 mL; 4:1). The combined filtrate was concentrated under reduced pressure to give compound 1, which was passed through a Sephadex LH-20 column using CH3OH-H2O (4:1) as eluant to give pure compound 1 (200 mg, 50 %). White powder; [α]D 25 + 17 (c 0.5, H2O); IR (KBr): 3436, 2944, 1609, 1378, 1144, 1092, 667 cm−1; 1H NMR (500 MHz, D2O): δ 5.26 (d, J = 3.5 Hz, 1 H, H-1D), 5.25 (d, J = 3.5 Hz, 1 H, H-1E), 5.06 (d, J = 3.5 Hz, 1 H, H-1B), 4.72 (d, J = 8.0 Hz, 1 H, H-1C), 4.60 (d, J = 8.0 Hz, 1 H, H-1A), 4.35–4.30 (m, 2 H, H-4C, H-5D), 4.29–4.24 (m, 2 H, H-5C, H-5E), 4.20 (dd, J = 10.5, 3.5 Hz, 1 H, H-2B), 4.15–4.05 (m, 3 H, H-2A, H-4B, OCH), 4.00–3.95 (m, 4 H, H-3B, H-3C, H-4A, OCH), 4.94–3.88 (m, 4 H, H-2C, H-2D, H-5A, H-5B), 3.85–3.73 (m, 10 H, H-2E, H-3A, H-4E, H-6abA, H-6abB, H-6abC, H-6aE), 3.70–3.65 (m, 3 H, H-3E, H-4D, H-6bE), 3.57 (dd, J = 10.0, 3.5 Hz, 1 H, H-3D), 3.32–3.20 (m, 2 H, NCH 2), 2.09, 2.06 (2 s, 6 H, 2 COCH 3), 1.20 (d, J = 6.5 Hz, 3 H, CCH 3); 13C NMR (125 MHz, D2O): δ 174.8, 173.8 (2 COCH3), 102.2 (C-1C), 100.9 (C-1A), 98.8 (C-1D), 92.9 (2 C, C-1B, C-1E), 76.0 (C-3B), 75.1 (C-4D), 74.6 (C-3C), 74.3 (C-3A), 74.2 (C-5A), 72.9 (C-5C), 71.8 (C-5E), 71.0 (C-2C), 70.9 (C-4E), 69.9 (C-5B), 69.4 (C-3E), 69.2 (C-2E), 68.8 (C-3D), 68.0 (C-2D), 67.7 (C-4A), 66.8 (C-5D), 65.7 (OCH2), 63.5 (C-4C), 63.4 (C-4B), 61.3 (C-6E), 61.1 (C-6C), 60.8 (C-6B), 60.7 (C-6A), 50.6 (C-2A), 48.8 (C-2B), 39.5 (NCH2), 22.3 (COCH3), 22.0 (COCH3), 15.3 (CCH3); MALDI-MS: 960.3 [M + Na]+; Anal. Calcd. for C36H63N3O25 (937.89): C, 46.10; H, 6.77 %; found: C, 61.50; H, 6.10 %.
Computational details
ROE based 2D ROESY NMR spectral analysis has been used to get detailed atomistic information of compound 1. Further, Mestro GUI of Desmond has been used for the preparation of bond optimization, configuration for α/β forms, L/D isomeric forms, and furanose/pyranose types of sugar rings in compound 1. Compound 1 was solvated using TIP3P water molecules [30] in a truncated octahedral box with edge distance of 10 Å. The account of non-bonded interaction was performed using a cutoff distance of 10 Å under isothermal-isobaric periodic boundary conditions. M-SHAKE algorithm was used for restraining all the hydrogen bonds with an integration time step of 2 fs [31]. Energy minimization, equilibration and production MD run for compound 1 were processed using OPLS_2005 force field in Desmond, which was already discussed in earlier reports [32, 33]. MD simulation was performed at 300 K for a time scale of 20 ns with recording an interval of 5 ps for trajectory frames.
References
Andersson D.I., Hughes D.: Persistence of antibiotic resistance in bacterial populations. FEMS Microbiol. Rev. 35, 901–911 (2011)
Bhatia S., Dimde M., Haag R.: Multivalent glycoconjugates as vaccines and potential drug candidates. Med. Chem. Commun. 5, 862–878 (2014)
Källenius G., Pawlowski A., Hamasur B., Svenson S.B.: Mycobacterial glycoconjugates as vaccine candidates against tuberculosis. Trends Microbiol. 16, 456–462 (2008)
Lucas A.H., Apicella M.A., Taylor C.E.: Carbohydrate moieties as vaccine candidates. Clin. Infect. Dis. 41, 705–712 (2005)
Astronomo R.D., Burton D.R.: Carbohydrate vaccines: developing sweet solutions to sticky situations? Nat. Rev. Drug Discov. 9, 308–324 (2010)
Lebeer S., Vanderleyden J., De Keersmaecker S.C.J.: Host interactions of probiotic bacterial surface molecules: comparison with commensals and pathogens. Nat. Rev. Microbiol. 8, 171–184 (2010)
Roberts I.S., Roberts I.S.: The biochemistry and genetics of capsular polysaccharide production in bacteria. Annu. Rev. Microbiol. 50, 285–315 (1996)
Raetz C.R.H., Whitfield C.: Lipopolysaccharide Endotoxins. Annu. Rev. Microbiol. 71, 635–700 (2002)
Nataro J.P., Kaper J.B.: Diarrheagenic Escherichia coli. Clin. Microbiol. Rev. 11, 142–201 (1998)
Andersson M., Carlin N., Leontein K., Lindquist U., Slettengren K.: Structural studies of the O-antigenic polysaccharide of Escherichia coli O86, which possesses blood-group B activity. Carbohydr. Res. 185, 211–223 (1989)
Yi W., Bystricky P., Yao Q., Guo H., Zhu L., Li H., Shen J., Li M., Ganguly S., Allen Bush C., Wang P.G.: Two different O-polysaccharides from Escherichia coli O86 are produced by different polymerization of the same O-repeating unit. Carbohydr. Res. 341, 100–108 (2006)
Niqudkar S.S., Demchenko A.V.: Stereocontrolled 1,2-cis glycosylation as the driving force of progress in synthetic carbohydrate chemistry. Chemical Sc. 6, 2687–2704 (2015)
Ingle A.B., Chao C.-S., Hung W.-C., Tony Mong K.-K.: Chemical synthesis of the O-antigen repeating unit of Escherichia coli O86 by an N-formylmorpholine-modulated one-pot glycosylation strategy. Asian J. Org. Chem. 3, 870–876 (2014)
Si A., Misra A.K.: Expedient synthesis of the pentasaccharide repeating unit of the polysaccharide O-antigen of Escherichia coli O11. Chemistry Open. 5, 47–50 (2016)
Santra A., Ghosh T., Misra A.K.: Expedient synthesis of two structurally close tetrasaccharides corresponding to the O-antigens of Escherichia coli O127 and Salmonella enterica O13. Tetrahedron-Asymmetry. 23, 1385–1392 (2012)
Lönn H.: Synthesis of a tri- and a hepta-saccharide which contain α-L-fucopyranosyl groups and are part of the complex type of carbohydrate moiety of glycoproteins. Carbohydr. Res. 139, 105–113 (1985)
Ghosh S., Misra A.K.: Concise synthesis of a hexasaccharide present in the cell wall lipopolysaccharide of Azospirillum lipoferum Sp59b. Tetrahedron-Asymmetry. 21, 725–730 (2010)
Ghosh S., Misra A.K.: Concise synthesis of a hexasaccharide related to the adhesin receptor of Streptococcus oralis ATCC 55229. J. Carbohydr. Chem. 28, 447–462 (2009)
Mukherjee, C., Misra, A. K. Glycosylation and pyranose-furanose isomerization of carbohydrates using HClO4-SiO2: synthesis of oligosaccharides containing galactofuranose. Synthesis 683–692 (2007).
Chakraborti A.K., Gulhane R.: Perchloric acid adsorbed on silica gel as a new, highly efficient, and versatile catalyst for acetylation of phenols, thiols, alcohols, and amines. Chem. Commun. 2003, (1896-1897)
Zemplén G.: Abbau der reduzierenden Biosen, I.: Direkte konstitutions-ermittlung der cellobiose. Ber. Dtsch. Chem. Ges. 59, 1254–1266 (1926)
Agnihotri G., Misra A.K.: Mild and efficient method for the cleavage of benzylidene acetals using HClO4-SiO2 and direct conversion of acetals to acetates. Tetrahedron Lett. 47, 3653–3658 (2006)
Demchenko, A., Stauch, T., Boons, G.-J. Solvent and other effects on the stereoselectivity of thioglycoside glycosidations. Synlett 818–820 (1997).
Satoh H., Hansen H.S., Manabe S., van Gunsteren W.F., Hünenberger P.H.: Theoretical investigation of solvent effects on glycosylation reactions: stereoselectivity controlled by preferential conformations of the intermediate oxacarbenium-counterion complex. J. Chem. Theory Comput. 6, 1783–1797 (2010)
Ogawa T., Yamamoto H.: Synthesis of a model linear mannohexaose for the backbone structure of fruit body polysaccharide of Tremella fuciformis and Dictyophora indusiata FISCH. Agric. Biol. Chem. 49, 475–482 (1985)
Nakahara Y., Ogawa T.: Solid-phase synthesis of an O-linked glycopeptide based on a benzyl-protected glycan approach. Carbohydr. Res. 292, 71–81 (1996)
Santra A., Ghosh T., Misra A.K.: Removal of benzylidene acetal and benzyl ether in carbohydrate derivatives using triethylsilane and Pd/C. Beilstein J. Org. Chem. 9, 74–78 (2013)
Santra A., Si A., Kar R.K., Bhunia A., Misra A.K.: Linear synthesis and conformational analysis of the pentasaccharide repeating unit of the cell wall O-antigen of Escherichia coli O13. Carbohydr. Res. 391, 9–15 (2014)
Kar R.K., Suryadevara P., Sahoo B.R., Sahoo G.C., Dikhit M.R., Das P.: Exploring novel KDR inhibitors based on pharmaco-informatics methodology. SAR QSAR Environ. Res. 24, 215–234 (2013)
Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L.: Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983)
Vincent K., van Gunsteren W.F., Hünenberger P.H.: A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 22, 501–508 (2001)
Dhara D., Kar R.K., Bhunia A., Misra A.K.: Convergent synthesis and conformational analysis of the hexasaccharide repeating unit of the O-antigen of Shigella flexneri serotype 1d. Eur. J. Org. Chem. 2014(21), 4577–4584 (2014)
Kar R.K., Suryadevara P., Jana J., Bhunia A., Chatterjee S.: Novel G-quadruplex stabilizing agents: in-silico approach and dynamics. J. Biomol. Struct. Dyn. 31, 1497–1518 (2013)
Acknowledgments
I. B. and R. K. K. thank CSIR, India for providing Senior Research Fellowships respectively. This work was supported by SERB, New Delhi, India (AKM) [Project No. EMR/2015/000282]. The authors sincerely thank reviewers for their valuable comments to improve the quality of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
ESM 1
(PDF 2869 kb)
Rights and permissions
About this article

Cite this article
Bhaumik, I., Kar, R.K., Bhunia, A. et al. Expedient synthesis of the pentasaccharide repeating unit of the O-antigen of Escherichia coli O86 and its conformational analysis. Glycoconj J 33, 887–896 (2016). https://doi.org/10.1007/s10719-016-9687-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10719-016-9687-x