
Abstract
The bisecting GlcNAc is transferred to the core mannose residue of complex or hybrid N-glycans on glycoproteins by the β1,4-N-acetylglucosaminyltransferase III (GlcNAcT-III) or MGAT3. The addition of the bisecting GlcNAc confers unique lectin recognition properties to N-glycans. Thus, LEC10 gain-of-function Chinese hamster ovary (CHO) cells selected for the acquisition of ricin resistance, carry N-glycans with a bisecting GlcNAc, which enhances the binding of the erythroagglutinin E-PHA, but reduces the binding of ricin and galectins-1, -3 and -8. The altered interaction with galactose-binding lectins suggests that the bisecting GlcNAc affects N-glycan conformation. LEC10 mutants expressing polyoma middle T antigen (PyMT) exhibit reduced growth factor signaling. Furthermore, PyMT-induced mammary tumors lacking MGAT3, progress more rapidly than tumors with the bisecting GlcNAc on N-glycans of cell surface glycoproteins. In recent years, evidence for a new paradigm of cell growth control has emerged involving regulation of cell surface residency of growth factor and cytokine receptors via interactions and cross-linking of their branched N-glycans with a lattice of galectin(s). Specific cross-linking of glycoprotein receptors in the lattice regulates their endocytosis, leading to effects on growth factor-induced signaling. This review will describe evidence that the bisecting GlcNAc of N-glycans regulates cellular signaling and tumor progression, apparently through modulating N-glycan/galectin interactions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Glycosylation is the most prevalent post-translational modification of membrane-bound and secreted glycoproteins that traverse the conventional secretory pathway. Most protein glycosylation is either Asn-linked or initiated by O-linked GalNAc added to Ser or Thr. The consensus site for N-glycans has recently been expanded to Phe–Yyy–Asn–Xxx–Thr in a Type I β-bulge and Phe–Yyy–Zzz–Asn–Xxx–Thr in a reverse turn (where Yyy can likely be any amino acid and Xxx is any amino acid but Pro) [1, 2]. The ability to predict O-GalNAc addition to Ser or Thr is improving based on in silico predictions [3, 4] and experimental determinations [5]. While all N-glycans have a common core consisting of Man3GlcNAc2Asn, there is currently no way of predicting the structures of the final complement of N-glycans on the many glycoforms of a glycoprotein. However, N-glycan structures may be determined by enzymatic release of N-glycans followed by glycomics analyses using mass spectrometry (MS) including matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) MS, gas chromatography (GC)/MS and tandem MS techniques [6]. Glycoproteomics is used to identify glycans at specific sites in a glycoprotein [7].
The bisecting GlcNAc is a unique modification of hybrid or complex N-glycans whose addition is catalyzed by β1,4-N-acetylglucosaminyltransferase III (GlcNAcT-III) or MGAT3 (E.C. 2.4.1.144) (Fig. 1a), an activity originally identified in hen oviduct [8]. In vitro glycosyltransferase assays indicated that the presence of a bisecting GlcNAc on a biantennary N-glycan terminating in GlcNAc prevents the subsequent action of N-glycan branching glycosyltransferases including GlcNAcT-II, GlcNAcT-IV, and GlcNAcT-V and the core fucosyltransferase FUT8 [9, 10]. Interestingly, however, glycomics profiling of the N-glycans from glycoproteins of LEC10 CHO cells that express the Mgat3 gene [11], revealed that many bisected N-glycans carry a core Fuc, and that N-glycans with up to 17 LacNAc units have a bisecting GlcNAc [12]. These LacNAc units must be extensions of branched N-glycans because LEC10 glycoproteins bind the lectins E-PHA and L-PHA much better than glycoproteins from parent CHO cells that do not express Mgat3 [13, 14]. Therefore, the inhibition of branching GlcNAc-transferases and FUT8 by the bisecting GlcNAc observed in vitro, does not occur in a CHO cell Golgi environment. Nevertheless, the bisecting GlcNAc profoundly affects the interaction of LEC10 cells with galactose-binding plant lectins including ricin [15, 16]. Thus, LEC10 CHO cells are highly resistant to ricin compared to parent CHO cells (Fig. 1b). By contrast, they are hypersensitive to the cytotoxicity of E-PHA and L-PHA, and bind more of these lectins than parent CHO cells. These data suggest that the bisecting GlcNAc has a major impact on the conformation of Gal residues in a bisected N-glycan. Models of N-glycans with and without a bisecting GlcNAc are consistent with this proposal [17, 18]. Thus, it is clear that the presence of the bisecting GlcNAc on the N-glycans of cell surface glycoproteins may modulate their interactions with galectins, siglecs or other glycan binding proteins. These effects may, however, vary with cell type because overexpression of the Mgat3 gene has been shown to reduce N-glycan branching or core fucosylation in some cell types (reviewed in [19, 20]).

The bisecting GlcNAc and lectin binding. a A proposed complex N-glycan containing the bisecting GlcNAc added by MGAT3 expressed in LEC10 cells, and the β1,6GlcNAc branch initiated by MGAT5 and absent from Lec4 mutant cells. b Lectin resistance test of CHO wild type and LEC10B cells expressing MGAT3 using the lectins ricin and E-PHA (adapted from [14]). c Flow cytometry of FITC-labeled galectin-3 binding to CHO, LEC10 or LEC11 cells in the presence or absence of lactose (courtesy of Santosh Patnaik [36]). LEC11 cells express Fut6 and add Fuc to LacNAc to generate the LeX and SLeX epitopes (Fig. 1a [76])
The Mgat3 gene has a unique tissue specific expression pattern with particularly high levels of transcripts in mouse brain and kidney, and a moderate level in intestine, based on Northern blot analyses [21, 22]. Kidney extracts are abundant in E-PHA-binding glycoproteins, consistent with the presence of the bisecting GlcNAc on N-glycans [14, 21]. Physiological functions of the bisecting GlcNAc have been proposed for the maintenance of kidney homeostasis [23]. However, mice with targeted inactivation of the Mgat3 gene, are viable and fertile with no gross anatomical or significant physiological abnormalities [22, 24] (http://www.functionalglycomics.org), suggesting that modification of N-glycans by the bisecting GlcNAc is dispensable for normal growth and development. Nevertheless, stress may reveal requirements that reflect predictions for functions of the bisecting GlcNAc. Interestingly, truncated, inactive MGAT3 produced by disruption of the Mgat3 gene by a neomycin cassette, causes a mild neurological phenotype in mice [13], suggesting that certain MGAT3 mutations in humans may have neurological or psychological effects.
In the past two decades, numerous studies have been directed towards understanding functions of the bisecting GlcNAc in modulating cell-cell and cell-matrix interactions, as well as cell growth control (reviewed in [19, 20, 25]). In this review, we focus on roles of the bisecting GlcNAc in galectin binding, growth factor signal transduction and tumor progression.
The bisecting GlcNAc and galectins
While the altered binding of bisected N-glycans to plant lectins is useful, an important question is whether the bisecting GlcNAc affects interactions of cell surface glycoproteins and endogenous animal lectins. Galectins belong to a large family of animal lectins that binds to β-galactosides. At least 15 galectins have been identified, although not all are found in every species [26, 27]. Galectins are expressed in the cytoplasm and nucleus and have been shown to play roles in intracellular regulation of pre-mRNA splicing [28]. However, many of their physiological functions in cell proliferation, survival, adhesion, migration and apoptosis have been attributed to their actions outside of the cell via glycan binding to cell surface glycoconjugates on cells, viruses or bacteria [29]. Lacking a signaling peptide, galectins are secreted via unconventional mechanism(s), which are poorly understood. However, some evidence suggests that both the glycan binding activity of a galectin and binding to ligand on the cell surface are required for efficient secretion [30, 31].
Galectins are categorized into three subtypes (proto, chimera, and tandem repeat) based on their sequence. All galectins contain at least one carbohydrate recognition domain (CRD) of ~130 amino acids, which interacts with glycans. Prototype galectins contain one CRD, and are normally present as a divalent homodimer. They include galectin-1, -2, -5, -7, -10, -11, -13, -14, and -15. Galectin-3 is the only chimera-type, with a CRD at the C-terminus and a long flexible N-terminus, which mediates oligomerization to form a pentameric structure that cross-links bound ligands [32]. Tandem-repeat type galectins include galectin-4, -6, -8, -9, -12. They are divalent with two distinct CRDs - one at the N-terminus and the other at the C-terminus connected by a linker peptide. Alternative splicing allows this linker region to be variable in length and may influence cross-linking potency [7, 32–34]. Furthermore, the presence of two CRDs with different glycan specificities may allow cross-linking of structurally distinct subsets of glycans [35, 36].
Complex N-glycans often carry repeating units of N-acetyl-lactosamine (LacNAc) Galβ1-4GlcNAc that bind several members of the galectin family [37]. The importance of LacNAc repeats for galectin binding to cell surfaces was shown using a series of CHO glycosylation mutants [36, 38]. Lec1 CHO mutants that lack hybrid and complex N-glycans on cell surface glycoproteins do not bind galectin-1, -3 or -8. Lec8 CHO mutants, that have no LacNAc units on N-glycans but carry terminal GalNAc on Ser/Thr residues, also bind very low amounts of these three galectins. Therefore, complex N-glycans are the major ligands for galectin-1, -3 and -8 on CHO cells. The effect of the bisecting GlcNAc on galectin binding was tested in LEC10 CHO cells in which most complex N-glycans carry the bisecting GlcNAc [12]. Binding of galectin-1, -3 and -8 was reduced to LEC10 compared to parent CHO cells [39]. The inhibitory effect of the bisecting GlcNAc for galectin binding was observed using fluorescinated galectins and flow cytometry (Fig. 1c; galectin-3) or using an array-based assay with glycans attached to a solid surface (Fig. 2; galectin-1). The effects of the bisecting GlcNAc on galectin binding have also been tested on chemoenzymatically synthesized bi-antennary N-glycans attached to BSA [18, 40]. Interestingly, the bisecting GlcNAc had no effect on the binding of galectin-4 to BSA-N-glycans, while the binding of galectin-1 was slightly enhanced and was further potentiated by the presence of core α1-6-linked fucose with the bisecting GlcNAc [18, 40]. By contrast, the binding of galectin-3 was reduced by the bisecting GlcNAc, consistent with its reduced binding to LEC10 CHO cells [39] (Fig. 1c). In another study, the promotion of laminin 332-dependent cell migration by galectin-3 in keratinocytes was inhibited when laminin 332 was modified with a bisected N-glycan [41], further supporting a negative effect of the bisecting GlcNAc on galectin-3 binding. It is thus clear, that the bisecting GlcNAc modulates galectin binding to complex N-glycans, and that it may therefore have functional consequences. It is also clear that care must be taken in extrapolating in vitro binding specificities to artificial substrates to specificities predicted for cell surface glycoproteins that may cluster and are mobile in the membrane [33].

Galectin-1 binding to CHO and glycosylation mutants. Mutant CHO cell lines (CHO, Lec1, LEC10, and Lec4) grown in monolayer or suspension culture were washed, biotinylated, harvested and fixed. The biotinylated cells (50,000 cells/well) were arrayed on neutravidin-coated black ELISA plates. A subset of biotinylated glycans from the Consortium for Functional Glycomics (CFG) Glycan Array version 2.3, including known binders and non-binders to galectin-1, were also arrayed on the same plate at a concentration of 60 pmol/well. Alexa-488 labeled human galectin-1 (30 μg/ml) was applied to each well in binding buffer (20 mm Tris–HCl, pH 7.4, 150 mm NaCl, 2 mm CaCl2, 2 mm MgCl2, and 0.05% Tween 20 with 1% bovine serum albumin (BSA)) and incubated for 1 h at room temperature. After galectin-1 removal, plates were washed three times with binding buffer lacking BSA and relative fluorescence units measured. Structures of complex N-glycans typical of CHO mutant cells and a subset of glycans on the array are shown. CHO cells contain β1,6GlcNAc branched complex N-glycans that lack the bisecting GlcNAc; Lec1 cells have no complex N-glycans; LEC10/LEC10B cells contain complex N-glycans modified with the bisecting GlcNAc; Lec4 cells lack both β1,6GlcNAc branched N-glycans and the bisecting GlcNAc. A typical Lec8 mutant complex N-glycan is shown as it is included in the model in Fig. 3. See Fig. 1a for glycan symbols. NC, no cells or compound; PBS, phosphate buffered saline (no galectin-1)
Complex N-glycans in signal transduction and growth control
Dennis and colleagues first reported that β1-6GlcNAc-branched complex N-glycans play important roles in growth control [42]. β1-6GlcNAc branching is initiated by the addition of GlcNAc to complex N-glycans by the enzyme GlcNAcT-V, encoded by the Mgat5 gene (Fig. 1a). When Mgat5 was ablated in transgenic mice overexpressing the polyoma middle T (PyMT) oncogene under the control of the mouse mammary tumor virus (MMTV) promoter, mammary tumor development and progression to lung metastasis were markedly reduced [42]. While the increase in β1-6GlcNAc-branched N-glycans had previously been noted in tumor formation [43], the report in 2000 was the first to present direct evidence suggesting that reducing the degree of N-glycan branching retards tumor progression. Importantly, a mechanism was subsequently proposed whereby branched N-glycans promote the interaction of growth factor/cytokine receptors (e.g., epidermal growth factor receptor (EGFR) and transforming growth factor-β (TGF-β) receptor) with galectin-3, thereby enhancing their cell surface residency time by preventing their loss due to constitutive endocytosis [44]. This in turn will increase the number of growth factor receptors on the cell surface that are available to respond to their respective ligands, leading to increased and prolonged ligand-induced signaling. This mechanism is supported by modeling studies [45] and investigations of autoimmunity modulated by surface residency of cytotoxic T-lymphocyte antigen 4 (CTLA4) [46, 47]. The retention of cell surface proteins by a galectin lattice has also been implicated in glucose homeostasis. For example, galectin-9 stabilizes glucose transporter 2 (GLUT2) on the cell surface increasing its half-life life in pancreatic β cells [48]. Reduced N-glycan branching due to the loss of MGAT4a or GLUT2 results in the development of diabetes [49]. Furthermore, the integrity of corneal epithelium that relies on cell surface signaling by vascular endothelial growth factor receptor 2 (VEGFR2) is dependent on interactions of branched N-glycans on VEGFR2 with galectin-3 [50]. In each of these cases, modification of a cell surface glycoprotein by highly branched N-glycans is essential to assure its optimal interactions with galectin(s). It should be noted, however, that roles for galectins in growth factor signaling may vary with cell context. Thus, the EGFR in human cancer cell lines with reduced MGAT5 activity and reduced N-glycan branching, does not exhibit enhanced ligand-induced endocytosis, but nevertheless shows impaired signal transduction that occurs primarily from endosomes in these cells [51]. The effect of branched N-glycans on growth factor signaling in this case appears to be galectin independent.
Galectin interactions with branched N-glycans may also function in regulating integrin-mediated cell motility. For example, galectin-3 stimulates α5β1 integrin-mediated activation of focal adhesion kinase (FAK) and phosphoinositide-3-kinase (PI3K) through its interactions with branched N-glycans of glycoproteins. This leads to increased fibronectin fibrillogenesis and fibronectin-dependent tumor cell spreading and motility, which may explain metastasis in MGAT5-expressing tumor cells [52]. α3β1 integrin is also activated by the interaction of galectin-3 with complex N-glycans to promote lamellipodia formation in corneal epithelial cells [53]. In general, cells with integrins carrying bisected branched N-glycans exhibit reduced migratory activity (reviewed in [19, 25, 54, 55]). In these cells, the addition of a bisecting GlcNAc concomitantly reduces β1-6GlcNAc branching, as indicated by reduced L-PHA binding to integrin subunits. This presumably reflects the fact that MGAT5 and other N-glycan branching glycosyltransferases may not utilize bisected N-glycans as acceptor substrates in certain cells [9]. Together, these studies suggest that the bisecting GlcNAc of complex or hybrid N-glycans may regulate biological functions of glycoproteins by altering N-glycan conformation, branching or composition, leading to reduced galectin binding.
The bisecting GlcNAc in growth factor signaling
Overexpression of Mgat3 in various cell lines has yielded valuable information on potential functions of MGAT3 in cell growth control and growth factor signaling. Bisected N-glycans are found on EGFR, which is often deregulated in cancer and plays key roles in the control of cell proliferation. In human U373 MG glioma cells, transfection of an Mgat3 cDNA causes reduced EGF binding and decreased EGFR autophosphorylation, but stimulates cell proliferation [56]. Overexpression of Mgat3 in Hela S3 cells increases signaling as shown by increased phosphorylation of extracellular signal-regulated kinase (ERK), and this correlates with reduced EGF binding, but increased EGFR endocytosis [57]. However, there is no change in EGFR dimerization or autophosphorylation in Mgat3-transfected Hela S3 cells. In PC2 neuronal cells, when Mgat3 is overexpressed, there is again a significant decrease in EGF binding and EGFR autophosphorylation, but this is accompanied by a decrease in ERK activation required for EGFR- and integrin-mediated neurite outgrowth [58]. Together, these results suggest that, while the bisecting GlcNAc of complex N-glycans affects EGFR-mediated signaling, the consequences vary with cell type, indicating that different mechanisms may underlie the effects of the bisecting GlcNAc on EGFR and other growth factor receptor signaling in different cellular environments.
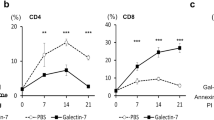
One caveat of experiments with transfected Mgat3 is that the overexpression of Mgat3 under a strong promoter may cause non-physiological effects. Therefore, LEC10 CHO mutants, that express Mgat3 from the endogenous CHO gene [11], provide a platform to study more physiological effects of the bisecting GlcNAc. Functions of the bisecting GlcNAc in cell growth control and growth factor signaling were compared between LEC10 CHO cells, wild type CHO cells that lack MGAT3 and bisected N-glycans, Lec4 CHO mutant cells lacking MGAT5 and β1-6GlcNAc-branched, as well as, bisected N-glycans, and Lec8 CHO mutant cells lacking LacNAc extensions of all N-glycan branches [39]. These cells were shown to bind galectins in the following order: CHO>LEC10>Lec4>Lec8. While these lines proliferate at a similar rate in medium containing 10% serum, each mutant grew slower than wild type in 7.5% serum [14]. To mimic conditions in PyMT-induced tumors, the CHO cells were transfected with PyMT. The growth rate of LEC10B/PyMT remained slower that that of CHO/PyMT cells, but slightly faster than Lec4/PyMT and Lec8/PyMT. To investigate growth factor signaling, the PyMT-expressing CHO lines were stimulated by platele-derived growth factor AB (PDGF-AB), since CHO cells express PDGFR but not EGFR. Responsiveness to PDGF-AB was significantly reduced in LEC10B/PyMT and Lec4/PyMT cells based on reduced ERK1/2 phosphorylation. For Lec8/PyMT cells, that do not bind galectins, there was no detectable response to PDGF-AB. When growth factor responsiveness was compared after treating the cells with lactose to remove surface-bound galectins, or sucrose as control, cells treated with lactose showed markedly reduced ERK1/2 activation, while sucrose treatment had no effect. Together, these results provide evidence for essential roles of growth factor receptor/galectin-lattice interactions in growth factor signaling in the CHO/PyMT lines. Therefore, it appears that the bisecting GlcNAc on N-glycans reduces growth factor signaling in a galectin-dependent manner (Fig. 3).

Model of galectin-dependent PDGFR signaling in CHO cells. Higher order clustering of PDGFRs on the CHO cell surface may be achieved through galectin-N-glycan interactions, which are thought to form a lattice that restrains endocytosis and promotes optimal ERK1/2 activation. Complex N-glycans on wild type CHO cells have the most binding sites for galectins and the greatest response to PDGF-AB. ERK1/2 activation is reduced in LEC10B cells that add the bisecting GlcNAc to complex N-glycans, and in Lec4 cells that lack a branch of complex N-glycans, and occurs at background levels in Lec8 cells that have few if any LacNAc units on complex N-glycans. Signaling strength correlates with the degree of interaction with the galectin lattice. Binding of N-glycans to galectin-3 pentamers is shown as an example, although other galectins are likely to participate in CHO galectin lattice(s). See Fig. 2 for complex N-glycan structures typical of CHO mutant cells
The bisecting GlcNAc in tumor progression
Evidence from cell-based assays and in vivo studies has shown that the bisecting GlcNAc may affect tumor progression and metastasis. In initial studies using tumor cell lines, overexpression of Mgat3 in a highly metastatic subclone of B16 melanoma cells resulted in significant suppression of lung colonization, which correlated with a decrease in β1-6GlcNAc branching [59]. By contrast, tumor growth and metastasis to spleen was increased in the same cell model, mediated in part by CD44-hyaluronan interactions enhanced by the bisecting GlcNAc on CD44 [60]. Furthermore, a K562 leukemia cell line overexpressing Mgat3 was resistant to natural killer cell cytotoxicity and showed increased spleen colonization [61]. Studies using transgenic mouse models expressing Mgat3 in liver also gave variable results.
In rat liver, Mgat3 expression is upregulated during chemically-induced hepatocarcinogenesis [62, 63], prompting an investigation into the function of Mgat3 in tumorigenesis induced by diethylnitrosamine (DEN). In mice overexpressing Mgat3 in liver under the control of the serum amyloid P component gene promoter, DEN-induced tumor incidence was significantly reduced [64]. On the other hand, no significant change in tumor incidence was observed in mice overexpressing Mgat3 under the control of the major urinary protein (MUP) promoter following treatment with DEN and phenobarbitol (PB)[65]. Additionally, tumor progression was retarded in mice with a targeted Mgat3 mutation after DEN alone or DEN and PB treatments [65, 66]. This appeared to be due to a non cell-autonomous mechanism since overexpression of Mgat3 in hepatocytes did not restore tumor progression to the levels obtained in wild type mice [65].
A direct role for the promotion of tumor progression by complex N-glycans has been established in mammary tumors induced by PyMT expressed from the MMTV/PyMT transgene [67]. In Mgat5 null mice that lack the β1-6GlcNAc branch of complex N-glycans (Fig. 1a), mammary tumor progression is greatly inhibited due to reduced growth factor signaling that can be restored by introduction of an Mgat5 cDNA into Mgat5 null tumor epithelial cells [44]. The consequence of MGAT5 deficiency in HER2-induced mammary tumorigenesis has also been investigated. As observed in the MMTV/PyMT transgenic model, mammary tumor cells lacking Mgat5 have reduced ERK1/2 and Akt/protein kinase B (PKB) activation [68]. Furthermore, in this case MGAT5 was implicated in tumor initiation and tumor onset. In humans, increased expression of complex N-glycans with β1-6GlcNAc branching is observed in breast and colorectal carcinomas, and the degree of expression correlates with the stage of progression of the cancer [69].
Because LEC10 CHO cells with the bisecting GlcNAc to N-glycans exhibit a reduced growth rate that persists when they overexpress the PyMT oncogene [14] and this correlates with reduced galectin-lattice dependent growth factor signaling, it was hypothesized that PyMT-induced mammary tumor progression might be enhanced in mice null for the Mgat3 gene. Mgat3 is not expressed in virgin mammary glands, however, its expression is upregulated during lactation and this is reflected by enhanced E-PHA binding to glycoproteins from mammary gland tissue lysates.
To investigate the effects of MGAT3 and the bisecting GlcNAc in tumor progression, MMTV/PyMT transgenic mice lacking MGAT3 were analyzed [14]. Mgat3 gene expression is observed in MMTV/PyMT mammary glands by around 4–5 weeks. Tumor onset and tumor sizes at 17 weeks are significantly enhanced in Mgat3 null mice. The first palpable tumor appears ~7 days earlier in mice lacking MGAT3. In addition, lung metastasis evaluated by the presence of PyMT transcripts in the lung, is significantly higher in Mgat3 null mice in the early stages of tumorigenesis, suggesting accelerated lung metastasis in the absence of MGAT3. Moreover, tumor cells isolated from Mgat3 null mice are more responsive to EGF- and PDGF-AB-induced growth factor signaling. Finally, an in vivo cell migration assay demonstrated that mammary tumor cells lacking MGAT3 have a higher migratory activity, with or without stimulation by EGF. Rescue of the Mgat3 null phenotype was evaluated by overexpressing Mgat3 in mammary gland under the control of MMTV promoter. Early-stage tumor development was delayed in the Mgat3 transgenic mice and they also exhibited reduced cell migratory activity in the in vivo cell migration assay. Thus, MGAT3 and the presence of the bisecting GlcNAc on mammary glycoproteins, reduces mammary tumor progression in a cell-autonomous manner. Based on the fact that the bisecting GlcNAc reduces galectin binding (Figs. 1 and 2), we propose that MGAT3 and the bisecting GlcNAc reduce galectin-lattice dependent growth factor signaling leading to retarded mammary tumor progression as depicted in the model in Fig. 3.
The galectin lattice
While galectins have been widely implicated as regulators of cell signaling [70], it remains unclear how galectin specificity is determined. The simple CHO cell synthesizes N-glycans with up to 26 LacNAc units [12], and all 15 galectins bind to LacNAc. The loss of one N-glycan branch and its associated LacNAc units, or the addition of the bisecting GlcNAc, reduces the binding of galectins, as discussed above. But how does this affect the nature of the predicted galectin lattice, which may include multiple galectins, depending on cell type? Thus, while the interaction of galectin-3 with growth factor receptors is proposed as part of the mechanism by which MGAT5 regulates growth factor signaling [44], genetic ablation of galectin-3 has no effect on mammary tumor progression [71]. If the enhancement of tumor growth and metastasis by MGAT5 were mediated solely by galectin-3, one would expect galectin-3 null mice to exhibit reduced tumor progression. It seems likely that one or more other galectins play a role in galectin-dependent cellular signaling, compensating for the absence of galectin-3. We have observed that eight galectin genes (galectin-1, -2, -3, -4, (−6), -7, -8, -9, and, -12) are expressed in mouse mammary tumor tissue (unpublished), and thus any combination, or all eight, could be involved in forming galectin lattices on the cell surface. In vitro titration experiments using concanavalin A have shown that structurally distinct and separable lattices are formed by Con A and Man5GlcNAc2 versus Con A and Man6GlcNAc2 [72]. Therefore, a range of intricate cross-linked lattices of different structure may potentially be formed by galectins and N-glycans, depending on fine glycan binding specificity, concentration and many other factors. Imaging such galectin lattices and determining the nature and specificity of their cross-linked structures is a key challenge for the future. Only then will it be possible to understand how the complement of galectins at the cell surface may function, alone or in concert, to control growth factor signaling, and how this control is altered by the presence of the bisecting GlcNAc on complex N-glycans.
Conclusions
In recent years, it has become apparent that complex N-glycans play pivotal roles in growth factor signaling and tumor progression. At the cell surface, the LacNAc units of N-glycans are cross-linked by galectins. We and others have shown that the bisecting GlcNAc on complex N-glycans modulates galectin interactions and thereby presumably affects galectin-lattice structure, the turnover of growth factor receptors and downstream signaling. Interestingly, the human MGAT3 gene is located on chromosome 22q13.2, in a region for which loss of heterozygosity has been associated with breast and colorectal cancer [73–75]. Thus, understanding the mechanisms by which MGAT3 and the bisecting GlcNAc alter growth factor signaling, tumor growth and metastasis may lead to prognostic or diagnostic assays for human cancers.
References
Culyba, E.K., Price, J.L., Hanson, S.R., Dhar, A., Wong, C.H., Gruebele, M., Powers, E.T., Kelly, J.W.: Protein native-state stabilization by placing aromatic side chains in N-glycosylated reverse turns. Science 331(6017), 571–575 (2011). doi:10.1126/science.1198461
Price, J.L., Powers, D.L., Powers, E.T., Kelly, J.W.: Glycosylation of the enhanced aromatic sequon is similarly stabilizing in three distinct reverse turn contexts. Proc Natl Acad Sci U S A 108(34), 14127–14132 (2011). doi:10.1073/pnas.1105880108
Gupta, R., Birch, H., Rapacki, K., Brunak, S., Hansen, J.E.: O-GLYCBASE version 4.0: a revised database of O-glycosylated proteins. Nucleic Acids Res 27(1), 370–372 (1999)
Nishikawa, I., Nakajima, Y., Ito, M., Fukuchi, S., Homma, K., Nishikawa, K.: Computational prediction of o-linked glycosylation sites that preferentially map on intrinsically disordered regions of extracellular proteins. Int J Mol Sci 11(12), 4991–5008 (2010). doi:10.3390/ijms11124991
Gerken, T.A., Jamison, O., Perrine, C.L., Collette, J.C., Moinova, H., Ravi, L., Markowitz, S.D., Shen, W., Patel, H., Tabak, L.A.: Emerging paradigms for the initiation of mucin-type protein O-glycosylation by the polypeptide GalNAc transferase family of glycosyltransferases. J Biol Chem 286(16), 14493–14507 (2011). doi:10.1074/jbc.M111.218701
North, S.J., Hitchen, P.G., Haslam, S.M., Dell, A.: Mass spectrometry in the analysis of N-linked and O-linked glycans. Curr Opin Struct Biol 19(5), 498–506 (2009). doi:10.1016/j.sbi.2009.05.005
Zielinska, D.F., Gnad, F., Wisniewski, J.R., Mann, M.: Precision mapping of an in vivo N-glycoproteome reveals rigid topological and sequence constraints. Cell 141(5), 897–907 (2010). doi:10.1016/j.cell.2010.04.012
Narasimhan, S.: Control of glycoprotein synthesis. UDP-GlcNAc:glycopeptide beta 4-N-acetylglucosaminyltransferase III, an enzyme in hen oviduct which adds GlcNAc in beta 1–4 linkage to the beta-linked mannose of the trimannosyl core of N-glycosyl oligosaccharides. J Biol Chem 257(17), 10235–10242 (1982)
Brockhausen, I., Narasimhan, S., Schachter, H.: The biosynthesis of highly branched N-glycans: studies on the sequential pathway and functional role of N-acetylglucosaminyltransferases I, II, III, IV, V and VI. Biochimie 70(11), 1521–1533 (1988)
Longmore, G.D., Schachter, H.: Product-identification and substrate-specificity studies of the GDP-L-fucose:2-acetamido-2-deoxy-beta-D-glucoside (FUC goes to Asn-linked GlcNAc) 6-alpha-L-fucosyltransferase in a Golgi-rich fraction from porcine liver. Carbohydr Res 100, 365–392 (1982)
Stanley, P., Sundaram, S., Tang, J., Shi, S.: Molecular analysis of three gain-of-function CHO mutants that add the bisecting GlcNAc to N-glycans. Glycobiology 15(1), 43–53 (2005). doi:10.1093/glycob/cwh136
North, S.J., Huang, H.H., Sundaram, S., Jang-Lee, J., Etienne, A.T., Trollope, A., Chalabi, S., Dell, A., Stanley, P., Haslam, S.M.: Glycomics profiling of Chinese hamster ovary cell glycosylation mutants reveals N-glycans of a novel size and complexity. J Biol Chem 285(8), 5759–5775 (2010). doi:10.1074/jbc.M109.068353
Bhattacharyya, R., Bhaumik, M., Raju, T.S., Stanley, P.: Truncated, inactive N-acetylglucosaminyltransferase III (GlcNAc-TIII) induces neurological and other traits absent in mice that lack GlcNAc-TIII. J Biol Chem 277(29), 26300–26309 (2002). doi:10.1074/jbc.M202276200
Song, Y., Aglipay, J.A., Bernstein, J.D., Goswami, S., Stanley, P.: The bisecting GlcNAc on N-glycans inhibits growth factor signaling and retards mammary tumor progression. Cancer Res 70(8), 3361–3371 (2010). doi:10.1158/0008-5472.CAN-09-2719
Campbell, C., Stanley, P.: A dominant mutation to ricin resistance in Chinese hamster ovary cells induces UDP-GlcNAc:glycopeptide beta-4-N-acetylglucosaminyltransferase III activity. J Biol Chem 259(21), 13370–13378 (1984)
Stanley, P., Caillibot, V., Siminovitch, L.: Selection and characterization of eight phenotypically distinct lines of lectin-resistant Chinese hamster ovary cell. Cell 6(2), 121–128 (1975)
Brisson, J.R., Carver, J.P.: Solution conformation of asparagine-linked oligosaccharides: alpha(1–2)-, alpha(1–3)-, beta(1–2)-, and beta(1–4)-linked units. Biochemistry 22(15), 3671–3680 (1983)
Andre, S., Unverzagt, C., Kojima, S., Frank, M., Seifert, J., Fink, C., Kayser, K., von der Lieth, C.W., Gabius, H.J.: Determination of modulation of ligand properties of synthetic complex-type biantennary N-glycans by introduction of bisecting GlcNAc in silico, in vitro and in vivo. Eur J Biochem/FEBS 271(1), 118–134 (2004)
Takahashi, M., Kuroki, Y., Ohtsubo, K., Taniguchi, N.: Core fucose and bisecting GlcNAc, the direct modifiers of the N-glycan core: their functions and target proteins. Carbohydr Res 344(12), 1387–1390 (2009). doi:10.1016/j.carres.2009.04.031
Stanley, P.: Biological consequences of overexpressing or eliminating N-acetylglucosaminyltransferase-TIII in the mouse. Biochim Biophys Acta 1573(3), 363–368 (2002)
Bhaumik, M., Seldin, M.F., Stanley, P.: Cloning and chromosomal mapping of the mouse Mgat3 gene encoding N-acetylglucosaminyltransferase III. Gene 164(2), 295–300 (1995)
Priatel, J.J., Sarkar, M., Schachter, H., Marth, J.D.: Isolation, characterization and inactivation of the mouse Mgat3 gene: the bisecting N-acetylglucosamine in asparagine-linked oligosaccharides appears dispensable for viability and reproduction. Glycobiology 7(1), 45–56 (1997)
Vagin, O., Tokhtaeva, E., Yakubov, I., Shevchenko, E., Sachs, G.: Inverse correlation between the extent of N-glycan branching and intercellular adhesion in epithelia. Contribution of the Na, K-ATPase beta1 subunit. J Biol Chem 283(4), 2192–2202 (2008). doi:10.1074/jbc.M704713200
Bhaumik, M., Harris, T., Sundaram, S., Johnson, L., Guttenplan, J., Rogler, C., Stanley, P.: Progression of hepatic neoplasms is severely retarded in mice lacking the bisecting N-acetylglucosamine on N-glycans: evidence for a glycoprotein factor that facilitates hepatic tumor progression. Cancer Res 58(13), 2881–2887 (1998)
Isaji, T., Kariya, Y., Xu, Q., Fukuda, T., Taniguchi, N., Gu, J.: Functional roles of the bisecting GlcNAc in integrin-mediated cell adhesion. Methods Enzymol 480, 445–459 (2010). doi:10.1016/S0076-6879(10)80019-9
Hirabayashi, J., Kasai, K.: The family of metazoan metal-independent beta-galactoside-binding lectins: structure, function and molecular evolution. Glycobiology 3(4), 297–304 (1993)
Houzelstein, D., Goncalves, I.R., Fadden, A.J., Sidhu, S.S., Cooper, D.N., Drickamer, K., Leffler, H., Poirier, F.: Phylogenetic analysis of the vertebrate galectin family. Mol Biol Evol 21(7), 1177–1187 (2004). doi:10.1093/molbev/msh082
Dagher, S.F., Wang, J.L., Patterson, R.J.: Identification of galectin-3 as a factor in pre-mRNA splicing. Proc Natl Acad Sci U S A 92(4), 1213–1217 (1995)
Di Lella, S., Sundblad, V., Cerliani, J.P., Guardia, C.M., Estrin, D.A., Vasta, G.R., Rabinovich, G.A.: When galectins recognize glycans: from biochemistry to physiology and back again. Biochemistry 50(37), 7842–7857 (2011). doi:10.1021/bi201121m
Seelenmeyer, C., Wegehingel, S., Tews, I., Kunzler, M., Aebi, M., Nickel, W.: Cell surface counter receptors are essential components of the unconventional export machinery of galectin-1. J Cell biol 171(2), 373–381 (2005). doi:10.1083/jcb.200506026
Seelenmeyer, C., Stegmayer, C., Nickel, W.: Unconventional secretion of fibroblast growth factor 2 and galectin-1 does not require shedding of plasma membrane-derived vesicles. FEBS Lett 582(9), 1362–1368 (2008). doi:10.1016/j.febslet.2008.03.024
Ahmad, N., Gabius, H.J., Andre, S., Kaltner, H., Sabesan, S., Roy, R., Liu, B., Macaluso, F., Brewer, C.F.: Galectin-3 precipitates as a pentamer with synthetic multivalent carbohydrates and forms heterogeneous cross-linked complexes. J Biol Chem 279(12), 10841–10847 (2004). doi:10.1074/jbc.M312834200
Earl, L.A., Bi, S., Baum, L.G.: Galectin multimerization and lattice formation are regulated by linker region structure. Glycobiology 21(1), 6–12 (2011). doi:10.1093/glycob/cwq144
Nishi, N., Itoh, A., Shoji, H., Miyanaka, H., Nakamura, T.: Galectin-8 and galectin-9 are novel substrates for thrombin. Glycobiology 16(11), 15C–20C (2006). doi:10.1093/glycob/cwl028
Carlsson, S., Oberg, C.T., Carlsson, M.C., Sundin, A., Nilsson, U.J., Smith, D., Cummings, R.D., Almkvist, J., Karlsson, A., Leffler, 12H.: Affinity of galectin-8 and its carbohydrate recognition domains for ligands in solution and at the cell surface. Glycobiology 17(6), 663–676 (2007). doi:10.1093/glycob/cwm026
Patnaik, S.K., Potvin, B., Carlsson, S., Sturm, D., Leffler, H., Stanley, P.: Complex N-glycans are the major ligands for galectin-1, -3, and −8 on Chinese hamster ovary cells. Glycobiology 16(4), 305–317 (2006). doi:10.1093/glycob/cwj063
Merkle, R.K., Cummings, R.D.: Asparagine-linked oligosaccharides containing poly-N-acetyllactosamine chains are preferentially bound by immobilized calf heart agglutinin. J Biol Chem 263(31), 16143–16149 (1988)
Cho, M., Cummings, R.D.: Galectin-1, a beta-galactoside-binding lectin in Chinese hamster ovary cells. II. Localization and biosynthesis. J Biol Chem 270(10), 5207–5212 (1995)
Patnaik, S.K., Stanley, P.: Lectin-resistant CHO glycosylation mutants. Methods Enzymol 416, 159–182 (2006). doi:10.1016/S0076-6879(06)16011-5
Andre, S., Kozar, T., Schuberth, R., Unverzagt, C., Kojima, S., Gabius, H.J.: Substitutions in the N-glycan core as regulators of biorecognition: the case of core-fucose and bisecting GlcNAc moieties. Biochemistry 46(23), 6984–6995 (2007). doi:10.1021/bi7000467
Kariya, Y., Kawamura, C., Tabei, T., Gu, J.: Bisecting GlcNAc residues on laminin-332 down-regulate galectin-3-dependent keratinocyte motility. J Biol Chem 285(5), 3330–3340 (2010). doi:10.1074/jbc.M109.038836
Granovsky, M., Fata, J., Pawling, J., Muller, W.J., Khokha, R., Dennis, J.W.: Suppression of tumor growth and metastasis in Mgat5-deficient mice. Nat Med 6(3), 306–312 (2000). doi:10.1038/73163
Dennis, J.W., Laferte, S., Waghorne, C., Breitman, M.L., Kerbel, R.S.: Beta 1–6 branching of Asn-linked oligosaccharides is directly associated with metastasis. Science 236(4801), 582–585 (1987)
Partridge, E.A., Le Roy, C., Di Guglielmo, G.M., Pawling, J., Cheung, P., Granovsky, M., Nabi, I.R., Wrana, J.L., Dennis, J.W.: Regulation of cytokine receptors by Golgi N-glycan processing and endocytosis. Science 306(5693), 120–124 (2004). doi:10.1126/science.1102109
Lau, K.S., Partridge, E.A., Grigorian, A., Silvescu, C.I., Reinhold, V.N., Demetriou, M., Dennis, J.W.: Complex N-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell 129(1), 123–134 (2007). doi:10.1016/j.cell.2007.01.049
Grigorian, A., Torossian, S., Demetriou, M.: T-cell growth, cell surface organization, and the galectin-glycoprotein lattice. Immunol Rev 230(1), 232–246 (2009). doi:10.1111/j.1600-065X.2009.00796.x
Demetriou, M., Granovsky, M., Quaggin, S., Dennis, J.W.: Negative regulation of T-cell activation and autoimmunity by Mgat5 N-glycosylation. Nature 409(6821), 733–739 (2001). doi:10.1038/35055582
Ohtsubo, K., Takamatsu, S., Minowa, M.T., Yoshida, A., Takeuchi, M., Marth, J.D.: Dietary and genetic control of glucose transporter 2 glycosylation promotes insulin secretion in suppressing diabetes. Cell 123(7), 1307–1321 (2005). doi:10.1016/j.cell.2005.09.041
Ohtsubo, K., Chen, M.Z., Olefsky, J.M., Marth, J.D.: Pathway to diabetes through attenuation of pancreatic beta cell glycosylation and glucose transport. Nat Med 17(9), 1067–1075 (2011). doi:10.1038/nm.2414
Markowska, A.I., Jefferies, K.C., Panjwani, N.: Galectin-3 protein modulates cell surface expression and activation of vascular endothelial growth factor receptor 2 in human endothelial cells. J Biol Chem 286(34), 29913–29921 (2011). doi:10.1074/jbc.M111.226423
Guo, H.B., Johnson, H., Randolph, M., Lee, I., Pierce, M.: Knockdown of GnT-Va expression inhibits ligand-induced downregulation of the epidermal growth factor receptor and intracellular signaling by inhibiting receptor endocytosis. Glycobiology 19(5), 547–559 (2009). doi:10.1093/glycob/cwp023
Lagana, A., Goetz, J.G., Cheung, P., Raz, A., Dennis, J.W., Nabi, I.R.: Galectin binding to Mgat5-modified N-glycans regulates fibronectin matrix remodeling in tumor cells. Mol Cell Biol 26(8), 3181–3193 (2006). doi:10.1128/MCB.26.8.3181-3193.2006
Saravanan, C., Liu, F.T., Gipson, I.K., Panjwani, N.: Galectin-3 promotes lamellipodia formation in epithelial cells by interacting with complex N-glycans on alpha3beta1 integrin. J Cell Sci 122(Pt 20), 3684–3693 (2009). doi:10.1242/jcs.045674
Zhao, Y., Sato, Y., Isaji, T., Fukuda, T., Matsumoto, A., Miyoshi, E., Gu, J., Taniguchi, N.: Branched N-glycans regulate the biological functions of integrins and cadherins. FEBS J 275(9), 1939–1948 (2008). doi:10.1111/j.1742-4658.2008.06346.x
Gu, J., Taniguchi, N.: Potential of N-glycan in cell adhesion and migration as either a positive or negative regulator. Cell Adhes Migrat 2(4), 243–245 (2008)
Rebbaa, A., Yamamoto, H., Saito, T., Meuillet, E., Kim, P., Kersey, D.S., Bremer, E.G., Taniguchi, N., Moskal, J.R.: Gene transfection-mediated overexpression of beta1,4-N-acetylglucosamine bisecting oligosaccharides in glioma cell line U373 MG inhibits epidermal growth factor receptor function. J Biol Chem 272(14), 9275–9279 (1997)
Sato, Y., Takahashi, M., Shibukawa, Y., Jain, S.K., Hamaoka, R., Miyagawa, J., Yaginuma, Y., Honke, K., Ishikawa, M., Taniguchi, N.: Overexpression of N-acetylglucosaminyltransferase III enhances the epidermal growth factor-induced phosphorylation of ERK in HeLaS3 cells by up-regulation of the internalization rate of the receptors. J Biol Chem 276(15), 11956–11962 (2001). doi:10.1074/jbc.M008551200
Gu, J., Zhao, Y., Isaji, T., Shibukawa, Y., Ihara, H., Takahashi, M., Ikeda, Y., Miyoshi, E., Honke, K., Taniguchi, N.: Beta1,4-N-Acetylglucosaminyltransferase III down-regulates neurite outgrowth induced by costimulation of epidermal growth factor and integrins through the Ras/ERK signaling pathway in PC12 cells. Glycobiology 14(2), 177–186 (2004). doi:10.1093/glycob/cwh016
Yoshimura, M., Nishikawa, A., Ihara, Y., Taniguchi, S., Taniguchi, N.: Suppression of lung metastasis of B16 mouse melanoma by N-acetylglucosaminyltransferase III gene transfection. Proc Natl Acad Sci U S A 92(19), 8754–8758 (1995)
Sheng, Y., Yoshimura, M., Inoue, S., Oritani, K., Nishiura, T., Yoshida, H., Ogawa, M., Okajima, Y., Matsuzawa, Y., Taniguchi, N.: Remodeling of glycoconjugates on CD44 enhances cell adhesion to hyaluronate, tumor growth and metastasis in B16 melanoma cells expressing beta1,4-N-acetylglucosaminyltransferase III. J Int Cancer 73(6), 850–858 (1997)
Yoshimura, M., Ihara, Y., Ohnishi, A., Ijuhin, N., Nishiura, T., Kanakura, Y., Matsuzawa, Y., Taniguchi, N.: Bisecting N-acetylglucosamine on K562 cells suppresses natural killer cytotoxicity and promotes spleen colonization. Cancer Res 56(2), 412–418 (1996)
Narasimhan, S., Schachter, H., Rajalakshmi, S.: Expression of N-acetylglucosaminyltransferase III in hepatic nodules during rat liver carcinogenesis promoted by orotic acid. J Biol Chem 263(3), 1273–1281 (1988)
Nishikawa, A., Fujii, S., Sugiyama, T., Hayashi, N., Taniguchi, N.: High expression of an N-acetylglucosaminyltransferase III in 3′-methyl DAB-induced hepatoma and ascites hepatoma. Biochem Biophys Res Commun 152(1), 107–112 (1988)
Ekuni, A., Miyoshi, E., Ko, J.H., Noda, K., Kitada, T., Ihara, S., Endo, T., Hino, A., Honke, K., Taniguchi, N.: A glycomic approach to hepatic tumors in N-acetylglucosaminyltransferase III (GnT-III) transgenic mice induced by diethylnitrosamine (DEN): identification of haptoglobin as a target molecule of GnT-III. Free Radic Res 36(8), 827–833 (2002)
Yang, X., Bhaumik, M., Bhattacharyya, R., Gong, S., Rogler, C.E., Stanley, P.: New evidence for an extra-hepatic role of N-acetylglucosaminyltransferase III in the progression of diethylnitrosamine-induced liver tumors in mice. Cancer Res 60(12), 3313–3319 (2000)
Yang, X., Tang, J., Rogler, C.E., Stanley, P.: Reduced hepatocyte proliferation is the basis of retarded liver tumor progression and liver regeneration in mice lacking N-acetylglucosaminyltransferase III. Cancer Res 63(22), 7753–7759 (2003)
Lau, K.S., Dennis, J.W.: N-Glycans in cancer progression. Glycobiology 18(10), 750–760 (2008). doi:10.1093/glycob/cwn071
Guo, H.B., Johnson, H., Randolph, M., Nagy, T., Blalock, R., Pierce, M.: Specific posttranslational modification regulates early events in mammary carcinoma formation. Proc Natl Acad Sci U S A 107(49), 21116–21121 (2010). doi:10.1073/pnas.1013405107
Fernandes, B., Sagman, U., Auger, M., Demetriou, M., Dennis, J.W.: Beta 1–6 branched oligosaccharides as a marker of tumor progression in human breast and colon neoplasia. Cancer Res 51(2), 718–723 (1991)
Boscher, C., Dennis, J.W., Nabi, I.R.: Glycosylation, galectins and cellular signaling. Curr Opin Cell Biol 23(4), 383–392 (2011). doi:10.1016/j.ceb.2011.05.001
Eude-Le Parco, I., Gendronneau, G., Dang, T., Delacour, D., Thijssen, V.L., Edelmann, W., Peuchmaur, M., Poirier, F.: Genetic assessment of the importance of galectin-3 in cancer initiation, progression, and dissemination in mice. Glycobiology 19(1), 68–75 (2009). doi:10.1093/glycob/cwn105
Mandal, D.K., Brewer, C.F.: Interactions of concanavalin A with glycoproteins: formation of homogeneous glycoprotein-lectin cross-linked complexes in mixed precipitation systems. Biochemistry 31(50), 12602–12609 (1992)
Castells, A., Gusella, J.F., Ramesh, V., Rustgi, A.K.: A region of deletion on chromosome 22q13 is common to human breast and colorectal cancers. Cancer Res 60(11), 2836–2839 (2000)
Iida, A., Kurose, K., Isobe, R., Akiyama, F., Sakamoto, G., Yoshimoto, M., Kasumi, F., Nakamura, Y., Emi, M.: Mapping of a new target region of allelic loss to a 2-cM interval at 22q13.1 in primary breast cancer. Gene Chromosome Canc 21(2), 108–112 (1998)
Nagahata, T., Hirano, A., Utada, Y., Tsuchiya, S., Takahashi, K., Tada, T., Makita, M., Kasumi, F., Akiyama, F., Sakamoto, G., Nakamura, Y., Emi, M.: Correlation of allelic losses and clinicopathological factors in 504 primary breast cancers. Breast cancer 9(3), 208–215 (2002)
Zhang, A., Potvin, B., Zaiman, A., Chen, W., Kumar, R., Phillips, L., Stanley, P.: The gain-of-function Chinese hamster ovary mutant LEC11B expresses one of two Chinese hamster FUT6 genes due to the loss of a negative regulatory factor. J Biol Chem 274(15), 10439–10450 (1999)
Acknowledgments
The authors gratefully acknowledge the contribution of Dr. Santosh Patnaik in acquiring the data presented in Fig. 1c. This work was supported by R01 CA30645 and R01 CA36434 to PS, training grant T32 CA009173-34 to HEM, and by resources from the Consortium for Functional Glycomics (Core D and Core H) funded by NIGMS 1U54 GM62116.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Miwa, H.E., Song, Y., Alvarez, R. et al. The bisecting GlcNAc in cell growth control and tumor progression. Glycoconj J 29, 609–618 (2012). https://doi.org/10.1007/s10719-012-9373-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10719-012-9373-6