
Abstract
Crabs feed on a wide range of items and display diverse feeding strategies. The primary objective of this study was to investigate 10 digestive enzyme genes in representative crabs to provide insights into the genetic basis of feeding habits among crab functional groups. Crabs were classified into three groups based on their feeding habits: herbivores (HV), omnivores (OV), and carnivores (CV). To test whether crabs’ feeding adaptations matched adaptive evolution of digestive enzyme genes, we examined the 10 digestive enzyme genes of 12 crab species based on hepatopancreas transcriptome data. Each of the digestive enzyme genes was compared to orthologous sequences using both nucleotide- (i.e., PAML and Datamonkey) and protein-level (i.e., TreeSAAP) approaches. Positive selection genes were detected in HV crabs (AMYA, APN, and MGAM) and CV crabs (APN, CPB, PNLIP, RISC, TRY, and XPD). Additionally, a series of positive selection sites were localized in important functional regions of these digestive enzyme genes. This is the first study to characterize the molecular basis of crabs’ digestive enzyme genes based on functional feeding group. Our data suggest that HV crabs have evolved an enhanced digestion capacity for carbohydrates, and CV crabs have acquired digestion capacity for proteins and lipids.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Crabs are one of the most species-rich groups among extant crustaceans, with over 7250 described species, and many crabs play an important role as a food source for humans with commercial value in fish markets worldwide (Wang et al. 2018c; De Grave et al. 2009). In the broadest sense, crabs could be classified into three functional feeding groups: herbivores (HV), omnivores (OV), and carnivores (CV). Many species of mangrove crabs, e.g., sesarmid crabs, are known to be herbivorous, and they can feed on mangrove pneumatophores, bark, and macroalgae (Dahdouh-Guebas et al. 1999; Wang et al. 2018a, 2019b). Portunid crabs are reported as being mainly carnivorous, preying on gastropods, crustaceans, bivalves, polychaetes, and sometimes fish (Choy 1986; Cannicci et al. 1996; Figueiredo and Anderson 2009). Yet most crabs are, to some degree, omnivorous, feeding on resources including aquatic plants, algae, plankton, molluscs, fish, worms, and organic matter (Figueiredo and Anderson 2009; Jin et al. 2003). As such, different crabs may have different feeding adaptations, yet little is known about the genetic basis of physiological adaptations.
Digestive enzyme activity is the most common indicator for evaluating the capacity for digestion (Dai et al. 2009). The digestion of food to obtain nutrients is a core physiological function (Wei et al. 2014), and digestive enzymes of animals with different feeding habits (herbivores, omnivores and carnivores) have different characteristics (Wang et al. 2016a, b). In crabs, the hepatopancreas is an important organ for the absorption and storage of nutrients, and can synthesize digestive enzymes for food digestion. Studies on hepatopancreatic secretions have identified trypsin, chymotrypsin, amylase, and lipase as the most important components for digestion in crustacean species (Fernández et al. 1997; Wei et al. 2014; Hammer et al. 2000). Despite hepatopancreas have important role in digestion (Figueiredo and Anderson 2009; Dammannagoda et al. 2015), research on digestive enzyme gene diversity and gene regulation in crabs’ hepatopancreas is rudimentary.
Next-generation sequencing technologies make it possible to generate large amounts of transcript sequences and gene expression data, including for non-model species without a sequenced genome (van Dijk et al. 2014; Zhu et al. 2017). In addition, de novo transcriptome provides great breadth and depth of information that can be used to allow cataloging of all genes expressed in a tissue, and facilitate detailed functional research regarding various proteins (Bain et al. 2016; Wang et al. 2018b). There are many transcriptomic analyses of crustaceans that help build a more complete understanding of regulatory mechanisms, such as heavy metal detoxication (Wang et al. 2019c), osmoregulation (Azam et al. 2016; Wang et al. 2018a), and oxidation resistance (Hui et al. 2017). However, transcriptomic profiling is thus far limited regarding molecular characteristics of crabs’ digestive enzyme genes.
In the present study, we performed adaptive evolution analysis on digestive enzyme protein-coding genes of 12 crab species based on hepatopancreas transcriptome data. We collected all the positively selected genes among CV and HV crabs, and compared them with a representative OV crab (Eriocheir japonica sinensis) to examine whether adaptive evolution is apparent. These results will reveal the genetic basis for some feeding adaptations of crabs, and improve our understanding of their genetic and evolutionary architecture.
Materials and methods
Sample collection, RNA extraction, and Illumina deep sequencing
The Illumina paired-end transcriptome data were generated from 12 crab species, including 3 CV, 4 HV, and 5 OV crabs (Table 1). All species were collected from a coastal mudflat wetland or surrounding waters in Shanghai, China. Biological information of the species is presented in Table S1. Crabs were placed in an ice bath for 1–2 min until anesthetized. The hepatopancreas was removed through surgery, immediately frozen in liquid nitrogen, and stored at − 80 °C until RNA extraction. All procedures followed guidelines for the care and use of laboratory animals from the Institutional Animal Care and Use Committee of Yancheng Teachers University, Yancheng, China. Total RNA was isolated from the hepatopancreas samples using Trizol reagent (Invitrogen, San Diego, CA, USA) following the manufacturer's protocol. The RNA quality and concentration were determined with a Nanodrop-2000 spectrophotometer (NanoDrop products, Thermo Fisher Scientific, Inc., Wilmington, DE, USA). Samples for whole transcriptome analysis were prepared using an Illumina kit (Illumina Inc., San Diego, CA, USA) following the manufacturer's recommendations. First, mRNA was purified using oligo (dT) magnetic beads, and the mRNA was split into short fragments (about 200 bp) using a fragmentation buffer. The first strand of cDNA was synthesized with random hexamer primers using the mRNA fragments as templates. Then, buffer, dNTPs, RNase H, and DNA polymerase I were added to the mixture to synthesize the second strand. Subsequently, sequencing adapters were ligated to the 5′ and 3′ ends of the fragments. The fragments were purified by agarose gel electrophoresis and enriched by PCR amplification to create a cDNA library. Eventually, the cDNA library was sequenced on an Illumina HiSeq X Ten platform (Illumina Inc.) and 100 bp paired-end reads were generated.
Transcriptome de novo assembly
Clean reads were obtained by removing reads containing an adaptor, reads containing poly-N (the ratio of ‘N’ to be more than 10%), and low quality reads (quality score < 20). Transcriptome assembly was accomplished based on clean reads using Trinity (Grabherr et al. 2011), with min_kmer_cov set to 2 and all other default parameters. The longest copy of redundant transcripts was regarded as a unigene. Unigenes were aligned to databases, including NR, String, Pfam, the Swiss-Prot, and the COG for eukaryotic complete genomes database, separately, using BLASTX with E values < 1E−5 (Altschul et al. 1997).
Identification of digestive enzyme genes and orthology inference
According to the annotation result, all unigenes related to most digestive enzymes were identified with the following criteria: the annotations of all-unigenes were obtained by a BLASTX (Camacho et al. 2009) search against the NR or Swiss-Prot database to match the corresponding digestive enzymes with E value < 1E−10. All candidate genes were selected manually and reconfirmed using the BLASTX network server in the National Center for Biotechnology Information (NCBI). The open reading frames (ORFs) of the putative digestive enzyme genes were predicted using an ORF finder (https://www.ncbi.nlm.nih.gov/orffinder/). In order to identify groups of putative orthologs, we adopted an approach based on sequence similarity and a tree-based approach (Yang and Smith 2014). Considered short sequences were not useful for the following analyses; only high quality and high integrity sequences were collected. Ultimately, 10 genes were chosen: Alpha-amylase (AMYA), N-aminopeptidase (APN), Carboxypeptidase B (CPB), Chymotrypsin-like proteinase (CTRL), Maltase-Glucoamylase (MGAM), Pancreatic lipase (PNLIP), Retinoid-inducible serine carboxypeptidase (RISC), Trypsin (TRY), Triacylglycerol lipase (TL), and xaa-Pro dipeptidase (XPD). All have a well-studied structure and function and are known enzymes in digesting lipid, protein, and carbohydrates. The detail functional information of the 10 genes is presented in Table S2, and the length of each gene is in Table S3. The sequences of the 10 digestive enzyme genes were verified by the genome data of Eriocheir japonica sinensis (PRJNA555707) and Portunus trituberculatus (PRJNA555262) (Tang et al, 2020a, b). The two species’ cDNA sequences are identical to the genomic data. Nucleotide sequences of each gene examined, and their deduced amino acid sequences, were aligned using MEGA 7.0 (Kumar et al. 2016) and manually adjusted with GeneDoc. The all sequences were deposited in GenBank under accession numbers MN964137–MN964248.
Adaptive evolution analysis
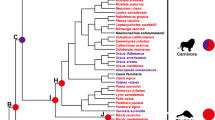
Estimating the nonsynonymous (dN)/synonymous substitution (dS) rate (ω = dN/dS) is considered a useful way to quantify the impact of natural selection on adaptive evolution (Ohta 1992; Wang et al. 2017, 2018a). The change of selective pressures can be indicated by the value of ω, where ω < 1, ω = 1, and ω > 1 correspond to purifying selection, neutral evolution, and positive selection, respectively (Wang et al. 2018a). Here, the selective pressure was tested based on phylogeny by using the codon-based maximum likelihood (CodeML) program in the PAML 4.7 package (Yang 2007). The well-supported phylogeny of Brachyura (Wang et al. 2019a; Shen et al. 2013) was used as the input tree in all analyses (Fig. 1).

A well-supported phylogeny of crabs used for selective pressure analysis in PAML. Positively selected genes identified in branch-site model are indicated
We first used the free-ratio and branch-site models (Zhang et al. 2005) implemented in CodeML to evaluate whether positive selection was restricted to specific HV or CV lineages. The free-ratio model (M1) assumes an independent ω ratio for each branch, and the one-ratio model (M0) which set up the same ω for all branches was used as the null hypothesis (Yang 1998). The improved branch-site model A (test 2) was applied to investigate the evolutionary rates of lineages of herbivores (HV), E. j. sinensis (OV), and carnivores (CV). Crabs of the three groups were respectively assigned as foreground, and compared with the null model, which assumes that all branches have the same evolutionary rate. The likelihood ratio test (LRT) statistic (2ΔL) approximates a chi-square distribution, and was used to test if there were significant evolutionary rate differences between foreground and background lineages. The Bayes empirical Bayes (BEB) approach (Yang et al. 2005) was used to identify amino acids under selection for CODEML.
Furthermore, to test for evidence of positive-selected sites in the digestive enzyme genes, we used the Datamonkey online server (https://www.datamonkey.org), that has the advantage of improving the dN/dS ratio estimate by incorporating variation in the rate of synonymous substitution (Yang et al. 2005). The internal fixed-effect likelihood (IFEL) and random-effect likelihood (REL), two different codon-based maximum likelihood methods, were used estimate the dN/dS ratio at every codon in the alignments (Pond and Frost 2005). The IFEL model estimates the ratio of dN/dS on a site-by-site basis, without assuming a priori distribution across sites. The REL model first fits a distribution of rates across sites and then infers the substitution rate for individual sites (Poon et al. 2009). Sites with p values < 0.1 for IFEL, and Bayes factor > 50 for REL, were considered as candidates under positive selection.
Finally, to further validate the result of PAML and Datamonkey, a complementary protein level approach was used in the TreeSAAP program, that compares non-synonymous residue property changes and identifies positive selection amino acid properties (Woolley et al. 2003). The TreeSAAP program detected sites based on 31 physicochemical amino acid properties, which were all magnitude category 6–8 changes; p values ≤ 0.01 were used as an index for the degree of radical amino acid substitution and positive selection (Wang et al. 2018a).
Mapping of positively selected sites onto protein structures
To gain insights into the functional significance of the putatively positive-selected sites, we mapped them onto the protein secondary and three-dimensional structures. We used the RaptorX (https://raptorx.uchicago.edu/) to predict the secondary structures of the implicated proteins, such as helix, beta-sheet, and coil (Källberg et al. 2014; Wang et al. 2016a, b). The protein structure domains were predicted by the PredictProtein web server (https://ppopen.rostlab.org/) (Yachdav et al. 2014). The 3D structures of genes under positive selection were predicted using the homology modeling software provided by the I-TASSER server (Zhang 2008). The radical amino sites under positive selection identified by more than one method were mapped onto the 3D structure using PYMOL.
Results
Transcriptome sequencing and de novo assembly
Obtaining cDNA sequences by transcriptome sequencing for evolutionary analysis has become an efficient option (Yuan et al. 2017; Ma et al. 2019). Here, the 12 newly generated crabs’ hepatopancreas transcriptomes contained a total of ~ 587.92 million clean reads after filtering, ranging from 38.38 to 57.10 million clean reads (Table 1). The assembled transcriptomes had an average of 228,925 transcripts with a N50 length of 307 to 1077 bp (Table 1). Number of predicted unigenes ranged from 113,860 to 399,653 per species, with an average length of 326.43 to 565.04 bp.
Molecular evolution of digestive enzyme genes in crabs
To test the selection constraints of different crab lineages for the 10 digestive enzyme genes, we used likelihood models of coding sequence evolution implemented in Codeml of the PAML package. With the one-ratio model (M0), that only allows a single ω ratio for all crab branches, the ω value of 10 genes ranged from 0.0695 to 0.270 (Table 2). The free-ratio (M1) model was significantly better than the M0 model (p < 0.05, Table 2) for eight genes (AMYA, APN, CPB, MGAM, PNLIP, RISC, TRY, XPD), suggesting heterogeneous selective pressures on the different lineages.
Then, we used the branch-site model to test for positive selection in individual codons for the three functional feeding groups (i.e., HV, OV, and CV). Considering that most crabs are omnivores, E. j. sinensis was set to represent OV group and the control for the other two groups. Results show that two (AMYA and MGAM) genes were identified under positive selection for HV, whereas no significant sign of positive selection was detected for CV (Table 3 and Fig. 1). Conversely, LRT tests showed that four genes (CPB, PNLIP, RISC, and XPD) were indicated as positive-selected in the ancestral branch of CV, but these genes were not detected as positive-selected in the HV group (Table 3 and Fig. 1). The ANY gene was determined to be positively selected in both HV and CV groups. 4 (HV), 2 (HV), 7(CV), 1(CV), 6 (CV), 2 (CV), and 38 (16 in HV, 22 in CV) sites were separately identified in AMYA, MGAM, CPB, PNLIP, RISC, XPD, and ANY, respectively, to be under selection using the BEB approach (posterior probabilities 0.60). Notably, no significant signs of positive selection for all 10 digestive enzyme genes were detected in the E. j. sinensis lineage (Table 3 and Fig. 1). These results are consistent with results drawn from free-ratio models.
Significant evidence of positive selection was further identified by the other two ML methods (IFEL and REL) implemented in Datamonkey, in which ω values were calculated based on the dS. The IFEL model results showed 33 positively selected sites in 7 genes (5 in AMYA, 9 in ANY, 4 in CPB, 8 in MGAM, 1 in PNLIP, 3 in RISC, and 3 in XPD) at a significance level < 0.1 (Table 4). In addition, REL also identified 11 codons in 3 genes (2 in AMYA, 6 in APN, and 3 in PNLIP) under positive selection at a level of Bayes factor > 50 (Table 4).
To support the ML method results, a complementary protein-level approach was implemented in TreeSAAP (Woolley et al. 2003). In TreeSAAP, the number of radical changes in the amino acid properties was used as a proxy for determining the strength of positive selection at a particular amino acid position (Sunagar et al. 2012). A total of 94 positively selected codons identified at three genes (17 in AMYA, 28 in MGAM, and 49 in APN) in the HV group (Table S4). Moreover, a series of putative positively selected sites from 5 osmoregulatory genes were identified in the CV group, i.e., 12 in APN, 19 in CPB, 14 in PNLIP, 14 in RISC, and 13 in XPD (Table S4).
Structural analyses of positively selected sites
To obtain insight into the functional significance of the putatively selected sites, we mapped all positively selected sites onto secondary structures of the corresponding digestive enzymes. It was found that the most of the positively selected sites were detected to fall in the regions of the functional regions within structures of the digestive enzymes (Table S5). One positively selected site in PNLIP (codon 267) was in the active site (Fig. 2).

Distribution of positively selected sites in the three-dimensional (3D) structure of PNLIP
Discussion
Animals are thought to be adaptive in their digestive enzyme production in response to differences in diet (German et al. 2004). Herbivorous animals normally exhibit higher carbohydrase activities (Horn 1989), and carnivorous animals frequently show higher lipid and protein enzyme activities (German et al. 2004; Hidalgo et al. 1999). Yet there is little data on the genetic basis underlying different feeding habits. To this end, we investigated selection pressure on the digestive enzyme genes of 12 crab species based on transcriptome data.
In the one-ratio model analysis, the ω values of all digestive enzyme genes were significantly less than 1, suggesting the general evolutionary pattern for crab digestive enzymes is conservative (Table 2). Even so, the free-ratio and branch-site model analyses still provide strong evidence that several digestive enzyme genes have been subjected to positive selection in HV and CV crabs (Tables 2, 3, and Fig. 1). According to previous studies, the positive selection signs are usually swamped by continuous negative selection that occurs on most sites in a gene sequence because positive selection mainly acts on only a few sites and for a short period of evolutionary time (Shen et al. 2010; Zhang et al. 2005). These reasons may partly explain why we can detect positive selection in some genes using the free-ratio and branch-site models, but not with the one-ratio model.
Selection analysis results showed significant evidence for positive selection at 7 of 10 digestive enzyme genes, i.e., AMYA (in HV), MGAM (in HV), CPB (in CV), PNLIP (in CV), RISC (in CV), XPD (in CV), and ANY (in HV and CV). Moreover, a series of sites were detected to be under positive selection in these genes using the codon-based maximum likelihood methods in Datamonkey (Table 4), which provides accessional evidence for positive selection. Adaptive evolution was also supported by evidence that the positively selected sites were identified by the protein-level approach in TreeSAAP (Table S4). Particularly, many of positively selected sites were localized on or near important structural regions (i.e., protein-binding region, alpha-helix, and beta-sheet) in predicted secondary and tertiary protein structures of the digestive enzyme, suggesting that these positively selected sites might have influenced protein properties and functions. Therefore, these nucleotide and protein-level analyses indicated positive selection may be a major driving force for evolution of digestive enzyme genes with respect to different functional feeding strategies.
Strong evidence for positive selection was noted at AMYA and MGAM in the HV group, in agreement with previous studies that carbohydrate-degrading enzymes (e.g., amylase, Maltase-Glucoamylase) tend to be higher in activity in guts of herbivorous crabs than in those of omnivores and carnivores (Dahdouh-Guebas et al. 1999). The AMYA gene encodes an alpha-amylase produced by the hepatopancreas. Alpha-amylases are common hydrolytic enzymes that break down polysaccharides by hydrolyzing alpha-d-(1,4)-glucan bonds, and thus catalyze the initial step in digestion of dietary starch, glycogen, and other related carbohydrates (Franco et al. 2000; Ramzi and Hosseininaveh 2010). The MGAM gene encodes maltase-glucoamylase, which is a brush-border membrane enzyme that plays a role in the final steps of digestion of starch. In addition, maltase-glucoamylase has an important role in herbivore evolution because of its importance in the breakdown of secondary metabolites of plants (Hemming and Lindroth 2000). Thus, the observed positive selection in AMYA and MGAM genes in HV crabs is suggestive of an enhanced capability for plant tissue digestion.
For CV crabs, lipids and proteins are the major nutritional components of food (Cannicci et al. 1996). Here, we detected positive selection of digestive enzyme genes involved with lipids and proteins might partly explain the molecular basis of CV crabs’ digestive adaptation mechanisms. PNLIP has a very important function in dietary lipids absorption by hydrolyzing triglycerides into diglycerides and subsequently into monoglycerides and free fatty acids (Mun et al. 2007). PNLIP only presented evidence of positive selection in CV crabs (Table 3), and most of positively selected sites in this gene were located primarily in functional domains that facilitated ligand-receptor interactions (Fig. 2). Particularly, codon 267 of PNLIP was in an active site (Fig. 2), which is hydrolyzed as the initial step in the activation process. Therefore, these positively selected amino acids in the key residues of PNLIP may have a positive impact on CV crabs’ lipid absorption ability. The detection of positive selection with PNLIP suggested that CV crabs may have acquired an enhanced capacity for lipid digestion. Three proteases (CPB, RISC, and XPD) were also only found to be under positive selection in CV crabs (Table 3), and they mainly play an important role in the intermediate step of protein digestion (Sakharov et al. 1997; Chen et al. 2001; Kumar et al. 2014). By contrast, proteases (CTRL and TRY) at the initiation stage of digestion (Perera et al. 2015) were not determined to be under positive selection (Table 3). These results suggested that CV crabs might have acquired an enhanced capacity for intermediate steps of protein digestion. In general, lipases and proteases subject to positive selection in CV crabs may be related with their complex diet and capabilities of digesting proteins and lipids.
Surprisingly, APN was determined to have undergone positive selection in both HV and CV crabs. APN (used as an indicator of protein digestive capacity) plays a major role in the final stages of dietary protein digestion in animals’ intestines, yielding various products, such as peptides which are finally digested by intracellular peptidases (Michiels et al. 2017; Tang et al. 2016). Thus, APN was consistent with evidence of positive selection in CV crabs and that these protease genes have important roles in enhancing digestion of protein. In comparison, positive selection identified in HV crabs seems to be more difficult to understand. In fact, herbivores also need a certain amount of plant protein from their food (Simpson et al. 2004). APN was also positively selected in HV crabs, suggesting that APN may play an important role in the digestion of plant protein. Further studies should be conducted regarding this interesting phenomenon to interpret roles of APN in HV crabs.
Conclusions
This study represents a preliminary survey of the molecular genetic basis underlying different feeding strategies in crabs. Significant positive selection genes were examined in HV crabs (carbohydrate digestive enzymes: AMYA and MGAM) and CV crabs (lipid digestive enzymes: PNLIP; protein digestive enzymes: CPB, RISC, and XPD). APN was detected with strong positive selection signals in both HV and CV crabs. Most of the putatively selected sites were localized in the important functional regions of these digestive enzyme genes. These results are consistent with the complex adaptations of crabs to digestion and absorption of diverse food resources.
References
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Azam M, Lifat RM, Tuan NV, Mather PB, Hurwood DA (2016) A transcriptomic scan for potential candidate genes involved in osmoregulation in an obligate freshwater palaemonid prawn (Macrobrachium australiense). PeerJ 4:e2520
Bain PA, Gregg AL, Kumar A (2016) De novo assembly and analysis of changes in the protein-coding transcriptome of the freshwater shrimp Paratya australiensis (Decapoda: Atyidae) in response to acid sulfate drainage water. BMC Genomics 17:1–18
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL (2009) BLAST+: architecture and applications. BMC Genomics 10:421
Cannicci S, Dahdouh-Guebas F, Dyane A, Vannini M (1996) Natural diet and feeding habits of Thalamita crenata (Decapoda: Portunidae). J Crustacean Biol 16:678–683
Chen J, Streb JW, Maltby KM, Kitchen CM, Miano JM (2001) Cloning of a novel retinoid-inducible serine carboxypeptidase from vascular smooth muscle cells. J Biol Chem 276:34175–34181
Choy SC (1986) Natural diet and feeding habits of the crabs Liocarcinus puber and L. holsatus (Decapoda, Brachyura, Portunidae). Mar Ecol Prog Ser 31:87–99
Dahdouh-Guebas F, Giuggioli M, Oluoch A, Vannini M, Cannicci S (1999) Feeding habits of non-ocypodid crabs from two mangrove forests in Kenya. Bull Mar Sci 64:291–297
Dai Y, Wang TT, Wang YF, Gong XJ, Yue CF (2009) Activities of digestive enzymes during embryonic development in the crayfish Procambarus clarkii (Decapoda). Aquac Res 40:1394–1399
Dammannagoda LK, Pavasovic A, Prentis PJ, Hurwood DA, Mather PB (2015) Expression and characterization of digestive enzyme genes from hepatopancreatic transcripts from redclaw crayfish (Cherax quadricarinatus). Aquac Nutr 21:868–880
De Grave S, Pentcheff ND, Ahyong ST, Chan T, Crandall KA, Dworschak PC, Felder DL, Feldmann RM, Fransen CH, Goulding LY (2009) A classification of living and fossil genera of decapod crustaceans. Raffles Bull Zool 21:1–109
Fernández I, Oliva M, Carrillo O, Wormhoudt AV (1997) Digestive enzyme activities of Penaeus notialis during reproduction and moulting cycle. Comp Biochem Physiol A Physiol 118:1267–1271
Figueiredo MSRB, Anderson AJ (2009) Digestive enzyme spectra in crustacean decapods (Paleomonidae, Portunidae and Penaeidae) feeding in the natural habitat. Aquac Res 40:282–291
Franco OL, Rigden DJ, Melo FR, Bloch C Jr, Silva CP, Grossi De Sá MF (2000) Activity of wheat α-amylase inhibitors towards bruchid α-amylases and structural explanation of observed specificities. Eur J Biochem 267(8):2166–2173
German DP, Horn MH, Gawlicka A (2004) Digestive enzyme activities in herbivorous and carnivorous prickleback fishes (Teleostei: Stichaeidae): ontogenetic, dietary, and phylogenetic effects. Physiol Biochem Zool 77:789–804
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29:644–652
Hammer HS, Bishop CD, Watts SA (2000) Activities of three digestive enzymes during development in the crayfish Procambarus clarkii (Decapoda). J Crustacean Biol 20:614–620
Hemming JD, Lindroth RL (2000) Effects of phenolic glycosides and protein on gypsy moth (Lepidoptera: Lymantriidae) and forest tent caterpillar (Lepidoptera: Lasiocampidae) performance and detoxication activities. Environ Entomol 29:1108–1115
Hidalgo MC, Urea E, Sanz A (1999) Comparative study of digestive enzymes in fish with different nutritional habits. Proteolytic and amylase activities. Aquaculture 170:267–283
Horn MH (1989) Biology of marine herbivorous fishes. Oceanogr Mar Biol 27:167–272
Hui BM, Song C, Liu Y, Li C (2017) Exploring the molecular basis of adaptive evolution in hydrothermal vent crab Austinograea alayseae by transcriptome analysis. PLoS ONE 12:e0178417
Jin G, Xie P, Li Z (2003) Food habits of two-year-old Chinese mitten crab (Eriocheir sinensis) stocked in Lake Bao'an, China. J Freshw Ecol 18:369–375
Källberg M, Margaryan G, Wang S, Ma J, Xu J (2014) RaptorX server: a resource for template-based protein structure modeling. Prot Struct Predict 1137:17–27
Kumar A, Are VN, Ghosh B, Agrawal U, Jamdar SN, Makde RD, Sharma SM (2014) Crystallization and preliminary X-ray diffraction analysis of Xaa-Pro dipeptidase from Xanthomonas campestris. Acta Crystallogr 70:1268–1271
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874
Ma K, Qin J, Lin C, Chan T, Ng P, Chu K, Tsang L (2019) Phylogenomic analyses of brachyuran crabs support early divergence of primary freshwater crabs. Mol Phylogenet Evol 135:62–66
Michiels MS, Del Valle JC, López Mañanes AA (2017) Trypsin and N-aminopeptidase (APN) activities in the hepatopancreas of an intertidal euryhaline crab: Biochemical characteristics and differential modulation by histamine and salinity. Comp Biochem Physiol A Mol Integr Physiol 204:228–235
Mun S, Decker EA, Mcclements DJ (2007) Influence of emulsifier type on in vitro digestibility of lipid droplets by pancreatic lipase. Food Res Int 40:770–781
Ohta T (1992) The nearly neutral theory of molecular evolution. Annu Rev Ecol Syst 23:263–286
Perera E, Rodríguez-Viera L, Perdomo-Morales R, Montero-Alejo V, Moyano FJ, Martínez-Rodríguez G, Mancera JM (2015) Trypsin isozymes in the lobster Panulirus argus (Latreille, 1804): from molecules to physiology. J Comp Physiol B 185:17–35
Pond SLK, Frost SD (2005) Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 21:2531–2533
Poon AF, Frost SD, Pond SLK (2009) Detecting signatures of selection from DNA sequences using Datamonkey. Methods Mol Biol 537:163–183
Ramzi S, Hosseininaveh V (2010) Biochemical characterization of digestive α-amylase, α-glucosidase and β-glucosidase in pistachio green stink bug, Brachynema germari Kolenati (Hemiptera: Pentatomidae). J Asia Pac Entomol 13:215–219
Sakharov DV, Plow EF, Rijken DC (1997) On the mechanism of the antifibrinolytic activity of plasma carboxypeptidase B. J Biol Chem 272:14477–14482
Shen Y, Liang L, Zhu Z, Zhou W, Irwin DM, Zhang Y (2010) Adaptive evolution of energy metabolism genes and the origin of flight in bats. Proc Natl Acad Sci USA 107:8666–8671
Shen H, Braband A, Scholtz G (2013) Mitogenomic analysis of decapod crustacean phylogeny corroborates traditional views on their relationships. Mol Phylogenet Evol 66:776–789
Simpson SJ, Sibly RM, Lee KP, Behmer ST, Raubenheimer D (2004) Optimal foraging when regulating intake of multiple nutrients. Anim Behav 68:1299–1311
Sunagar K, Johnson WE, O'Brien SJ, Vasconcelos V, Antunes A (2012) Evolution of CRISPs associated with toxicoferan-reptilian venom and mammalian reproduction. Mol Biol Evol 29:1807–1822
Tang B, Zhang D, Li H, Jiang S, Zhang H, Xuan F, Ge B, Wang Z, Liu Y, Sha Z, Cheng Y, Jiang W, Jiang H, Wang Z, Wang K, Li C, Sun Y, She S, Qiu Q, Wang W, Li X, Li Y, Liu Q (2020a) Chromosome-level genome assembly reveals the unique genome evolution of the swimming crab (Portunus trituberculatus). GigaScience 9(1):161
Tang B, Wang Z, Liu Q, Zhang H, Jiang S, Li X, Wang Z, Sun Y, Sha Z, Jiang H, Wu X, Ren Y, Li H, Xuan F, Ge B, Jiang W, She S, Sun H, Qiu Q, Wang W, Wang Q, Qiu G, Zhang D, Li Y (2020b) High-quality genome assembly of Eriocheir japonica sinensis reveals its unique genome evolution. Front Genet 10:1340
Tang J, Qu F, Tang X, Zhao Q, Wang Y, Yi Z, Feng J, Lu S, Hou D, Zhen L (2016) Molecular characterization and dietary regulation of aminopeptidase N (APN) in the grass carp (Ctenopharyngodon idella). Gene 582:77–84
Van Dijk EL, Auger H, Jaszczyszyn Y, Thermes C (2014) Ten years of next-generation sequencing technology. Trends Genet 30:418–426
Wang S, Peng J, Ma J, Xu J (2016a) Protein secondary structure prediction using deep convolutional neural fields. Sci Rep UK 6:18926–18926
Wang Z, Xu S, Du K, Fang H, Zhuo C, Zhou K, Ren W, Yang G (2016b) Evolution of digestive enzymes and RNASE1 provides insights into dietary switch of cetaceans. Mol Biol Evol 33:3144–3157
Wang Z, Shi X, Sun L, Bai Y, Zhang D, Tang B (2017) Evolution of mitochondrial energy metabolism genes associated with hydrothermal vent adaption of Alvinocaridid shrimps. Genes Genomics 39:1367–1376
Wang Z, Bai Y, Zhang D, Tang B (2018a) Adaptive evolution of osmoregulatory-related genes provides insight into salinity adaptation in Chinese mitten crab, Eriocheir sinensis. Genetica 146:303–311
Wang Z, Sun L, Guan W, Zhou C, Tang B, Cheng Y, Huang J, Xuan F (2018b) De novo transcriptome sequencing and analysis of male and female swimming crab (Portunus trituberculatus) reproductive systems during mating embrace (stage II). BMC Genet 19:3
Wang Z, Wang Z, Shi X, Wu Q, Tao Y, Guo H, Ji C, Bai Y (2018c) Complete mitochondrial genome of Parasesarma affine (Brachyura: Sesarmidae): Gene rearrangements in Sesarmidae and phylogenetic analysis of the Brachyura. Int J Biol Macromol 118:31–40
Wang Z, Shi X, Guo H, Tang D, Bai Y, Wang Z (2019a) Characterization of the complete mitochondrial genome of Uca lacteus and comparison with other Brachyuran crabs. Genomics 112:10–19
Wang Z, Shi X, Tao Y, Wu Q, Bai Y, Guo H, Tang D (2019b) The complete mitochondrial genome of Parasesarma pictum (Brachyura: Grapsoidea: Sesarmidae) and comparison with other Brachyuran crabs. Genomics 111:799–807
Wang Z, Tang D, Sun L, Shi X, Liu R, Guo H, Tang B (2019c) Comparative transcriptome analysis in the hepatopancreas of Helice tientsinensis exposed to the toxic metal cadmium. Genes Genomics 41(4):417–429
Wei J, Zhang X, Yu Y, Li F, Xiang J (2014) RNA-Seq reveals the dynamic and diverse features of digestive enzymes during early development of Pacific white shrimp Litopenaeus vannamei. Comp Biochem Physiol Part D Genomics Proteomics 11:37–44
Woolley S, Johnson J, Smith MJ, Crandall KA, McClellan DA (2003) TreeSAAP: selection on amino acid properties using phylogenetic trees. Bioinformatics 19:671–672
Yachdav G, Kloppmann E, Kajan L, Hecht M, Goldberg T, Hamp T, Hönigschmid P, Schafferhans A, Roos M, Bernhofer M (2014) PredictProtein—an open resource for online prediction of protein structural and functional features. Nucleic Acids Res 42:337–343
Yang Z (1998) Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol Biol Evol 15:568–573
Yang Z (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24:1586–1591
Yang Y, Smith SA (2014) Orthology inference in nonmodel organisms using transcriptomes and low-coverage genomes: improving accuracy and matrix occupancy for phylogenomics. Mol Biol Evol 31:3081–3092
Yang Z, Wong WS, Nielsen R (2005) Bayes empirical bayes inference of amino acid sites under positive selection. Mol Biol Evol 22:1107–1118
Yuan J, Zhang X, Liu C, Duan H, Li F, Xiang J (2017) Convergent evolution of the osmoregulation system in decapod shrimps. Mar Biotechnol 19:76–88
Zhang Y (2008) I-TASSER server for protein 3D structure prediction. BMC Genomics 9:40
Zhang J, Nielsen R, Yang Z (2005) Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol Biol Evol 22:2472–2479
Zhu R, Liu X, Lv X, Li S, Li Y, Yu X, Wang X (2017) Deciphering transcriptome profile of the yellow catfish (Pelteobagrus fulvidraco) in response to Edwardsiella ictaluri. Fish Shellfish Immunol 70:593–608
Funding
This study was funded by the National Natural Science Foundation of China (grant number 31702014), and Doctoral Scientific Research Foundation of Yancheng Teachers University to ZFW, and Open Foundation of Jiangsu Key Laboratory for Bioresources of Saline Soils (grant number JKLBS2019006).
Author information
Authors and Affiliations
Contributions
ZFW, DT, and HYG designed and conceived the experiment. ZFW, CCS, LW, and YQL performed the data analysis and draft the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The all authors declare there are no competing interests.
Ethical approval
The sampling location was not privately-owned or protected, and field sampling did not involve protected species.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
10709_2020_90_MOESM5_ESM.xlsx
Supplement Table S5 Secondary structure of digestive enzyme depicting the locations of positively selected amino acids detected by 3 methods (PAML,Datamonky and TreeSAAP) (XLSX 36 kb)
Rights and permissions
About this article

Cite this article
Wang, Z., Tang, D., Guo, H. et al. Evolution of digestive enzyme genes associated with dietary diversity of crabs. Genetica 148, 87–99 (2020). https://doi.org/10.1007/s10709-020-00090-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10709-020-00090-7