
Abstract
Satellite DNA (satDNA) is a non-coding component of eukaryotic genomes, located mainly in heterochromatic regions. Relevance of satDNA began to emerge with accumulating evidence of its potential yet hardly comprehensible role that it can play in the genome of many organisms. We isolated the first satDNA of the codling moth (Cydia pomonella, Tortricidae, Lepidoptera), a species with holokinetic chromosomes and a single large heterochromatic element, the W chromosome in females. The satDNA, called CpSAT-1, is located on all chromosomes of the complement, although in different amounts. Surprisingly, the satellite is almost missing in the heterochromatic W chromosome. Additionally, we isolated mRNA from all developmental stages (1st–5th instar larva, pupa, adult), both sexes (adult male and female) and several tissues (Malpighian tubules, gut, heart, testes, and ovaries) of the codling moth and showed the CpSAT-1 sequence was transcribed in all tested samples. Using CpSAT-1 specific primers we amplified, cloned and sequenced 40 monomers from cDNA and gDNA, respectively. The sequence analysis revealed a high mutation rate and the presence of potentially functional motifs, mainly in non-conserved regions of the monomers. Both the chromosomal distribution and the sequence analysis suggest that CPSAT-1 has no function in the C. pomonella genome.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Satellite DNA (satDNA) is a significant component of genomic DNA (gDNA) in eukaryotic organisms (Charlesworth et al. 1994). It consists of tandem repeats organized in long uninterrupted arrays, which can be recognized as a ladder of bands on an electrophoretic gel after digestion of gDNA with proper restriction endonuclease. The satellite arrays are composed of repeated units (the monomers) organized in head-to-tail manner and their length can reach up to 100 Mbp (Plohl et al. 2008, and references therein). The monomer length and composition are highly variable and reach from simple several tens bp long sequence to complex higher level structures with internal repeats and open reading frames whose length exceeds 1 kbp (reviewed by Palomeque and Lorite 2008).
Although satellite DNA is usually considered to be junk DNA, growing body of evidence suggests that at least some satellites perform important functions for the host genome. Typically, satDNAs often occur in the centromeres of eukaryotic chromosomes where they are necessary for their proper function. Similarly, the satDNA was found in telomeres in some chironomids and the mosquito Anopheles gambiae (Diptera) which lost the typical arthropod telomeric sequence (TTAGG)n and maintain their telomeres via crossing-over between satDNA stretches (Saiga and Edström 1985; Biessmann et al. 1996). Finally, satDNA can influence gene expression, such as TCAST1 in the beetle Tribolium castaneum, which has been shown to suppress the expression of various protein-coding genes after heat shock (Feliciello et al. 2015). SatDNAs performing a structural or functional role may contain various functional motifs, such as binding sites for centromeric proteins (e.g. Masumoto et al. 1989), promoters for RNA polymerase II and III (e.g. Renault et al. 1999), or transcription factor binding sites (e.g. Metz et al. 2004). However, RNA transcripts of some satDNAs have been shown to be equally important. For example, in Schizosaccharomyces pombe the small interfering RNA (siRNA) derived from the centromeric satellite sequence is involved in the RNAi machinery and it is crucial for local heterochromatin assembly as well, thus ensuring the proper centromere function (Volpe et al. 2002). Transcripts of the alphoid satellite DNA in zebrafish and chicken are tissue and time specific, and they probably regulate the expression of various genes whose mRNAs contain alphoid-like sequence in their UTR regions (Li and Kirby 2003). In hymenopteran insects, some satDNA transcripts are instar, sex or caste specific, which, again, suggests their regulatory role (Rouleux-Bonnin et al. 1996; Lorite et al. 2002; Rouleux-Bonnin et al. 2004). Finally, satDNA transcripts in Schistosoma blood-flukes and cricket have been shown to possess the self-cleavage ability, although their role remains to be resolved (Ferbeyre et al. 1998).
SatDNA is found mainly in heterochromatic regions, such as centromeres, subtelomeres and Y or W sex chromosomes (Charlesworth et al. 1994; Palomeque and Lorite 2008). Most information about satDNA comes from organisms with monocentric chromosomes, while only limited data are available from species with holokinetic chromosomes, i.e. chromosomes lacking one of the most frequent locations of satDNA, the centromere. The most comprehensive study of satDNA variability and distribution in an individual species with holokinetic chromosomes was performed in the woodrush Luzula elegans (Heckmann et al. 2013). The authors identified thirty-seven families of satellite repeats, together forming nearly 10 % of the L. elegans genome. Analysis of the distribution of the most abundant satellites revealed that these tandem repeats tend to form distinct bands located mainly in the subtelomeric regions. Importantly, none of the satellites showed chromosome-wide distribution, which would be expected in the holokinetic chromosomes. In insects, the satDNA was found in several representatives of three orders with holokinetic chromosomes: Lepidoptera (moths and butterflies), Homoptera (aphids) (reviewed by Palomeque and Lorite 2008), and Heteroptera (true bugs) (Bardella et al. 2014). Although the number of satDNAs isolated from insect species with holokinetic chromosomes is rather low, they managed to provide quite inconsistent data. Naturally, they prefer location in heterochromatic regions, such as the subtelomeric chromosome segments in aphids and the kissing bug Triatoma infestans (Spence et al. 1998; Bardella et al. 2014) or the W chromosome in lepidopteran females (Lu et al. 1994; Mandrioli et al. 2003). However, the frequent targets are also the X and Z chromosomes (Bizzaro et al. 1996; Mandrioli et al. 1999, 2003; Bardella et al. 2014). Finally, the satDNA AmTFR from Antheraea mylitta (Lepidoptera) was shown to be dispersed on all chromosomes (Mahendran et al. 2006).
Despite the large-scale genome sequencing, satDNA remains rather neglected part of the genomes due to its location in heterochromatin and the fact that its long arrays composed of repetitive sequences resist to subsequent sequence assembly. In order to search for new satDNAs, we performed restriction digestion of gDNA of the codling moth Cydia pomonella, a lepidopteran species with holokinetic chromosomes with the only visible heterochromatic region represented by the entire female sex-determining W chromosome (Fuková et al. 2005, 2007). We believed that this approach could reveal a satDNA with either curious pattern of location or, more interestingly, a functional satellite sequence. In this study, we present a new satDNA located on every chromosome of the complement and transcribed in all developmental stages of the codling moth.
Materials and methods
Insects
We used a laboratory strain (Krym-61) of the codling moth strain which is maintained at the Institute of Entomology, BC CAS in České Budějovice since 2002. Details about its origin, artificial diet, and rearing conditions are given in Fuková et al. (2005).
Restriction analysis of C. pomonella gDNA and cloning of restriction fragments
Total genomic DNA (gDNA) was extracted by standard phenol–chloroform-isoamylalcohol procedure from adult females of the codling moth (Ausubel et al. 2003). 5–10 μg of gDNA were digested with 10–15 U of respective restriction endonuclease (RE) at 37 °C overnight. Restriction products were separated electrophoretically on 1.5 % agarose gel in TAE buffer (Online Resource 1). For further analysis we selected restriction products of XbaI RE which provided four bands (100, 150, 350, and 500 bp long). Bands of interest were cut out under UV light, their DNA was isolated with the Wizard SV Gel and PCR-up System (Promega Corporation, Madison, WI, USA) and cloned into the pUC19 vector digested with XbaI RE. The ligation reaction contained 50 ng of the vector, 11–25 ng of the DNA, 1× buffer and T4 DNA ligase (TaKaRa, Otsu, Japan). Ligation reaction was incubated overnight at 16 °C. The product of ligation reaction was used for heat-shock transformation of chemically competent DH5α cells of Escherichia coli.
Probes for Southern hybridization and FISH
CpSAT1 DIG- or biotin-labelled probes for Southern hybridization and fluorescence in situ hybridization (FISH), respectively, were generated by means of PCR from pUC19 plasmids containing inserted satellite monomers. 12.5 µl reaction mix contained 1× Ex Taq buffer, dNTP mix (0.35 mM DIG-dUTP or 0.35 mM biotin-dUTP (both Roche Diagnostics, Basel, Switzerland), 1 mM dGTP, dCTP, dATP, and 0.65 mM dTTP), M13-24 and M13-26 primers (6 µM of each), 0.5 U of Ex Taq Hot Start DNA polymerase (TaKaRa), and 10 ng of DNA template. PCR profile was as follows: predenaturation at 94 °C for 3 min, denaturation at 94 °C for 30 s, annealing at 57 °C for 30 s, elongation at 72 °C for 60 s (steps 2–4 were repeated 30 times), postelongation at 72 °C for 7 min.
Southern hybridization
Total gDNA was extracted as described above. 3 μg of gDNA were digested with 3 U of XbaI at 37 °C for 1 h. Restriction products were separated on 1 % agarose gel in TBE buffer. Southern hybridization was performed according to Traut et al. (2007). The hybridization experiment was repeated twice.
Chromosome preparations
Mitotic chromosomes were obtained from wing imaginal discs of the 5th instar female larvae of C. pomonella. Pachytene chromosomes were obtained from ovaries of early female pupae. Preparations were made according to a slightly modified procedure of Sahara et al. (1999). The organs were dissected in physiological solution. The wing discs were hypotonized in 0.075 M KCl for 10 min, then fixed in Carnoy fixative (100 % ethanol-chloroform–acetic acid, 6:3:1) for 15 min. The ovaries were fixed without hypotonization. After fixation the organs were transferred on a slide into a drop of 60 % acetic acid and macerated. Finally, the material was spread on a histological plate heated to 45 °C. Then the chromosome preparations were dehydrated in an ethanol series (70, 80, and 100 %, 30 s each) and stored at −20 °C until further use.
FISH
FISH with biotinylated probe was performed essentially following the procedure in Sahara et al. (1999) with some modifications including those described in Fuková et al. (2005). Briefly, after removal from the freezer, dehydration in the ethanol series (see above) and air-drying, the slides were baked for 30 min at 60 °C, treated with RNase A (Sigma-Aldrich, St. Louis, MO, USA) (20 μg in 100 μL 2× SSC) for 1 h, washed twice in 2× SSC for 5 min, then treated with proteinase K (Sigma-Aldrich) (100 μg in 100 mL of PBS) for 5 min, washed twice in 2× SSC for 5 min, and finally incubated in 5x Denhardt’s solution for 30 min. All incubations and washes were performed at 37 °C. Denaturation of chromosomes was done at 68 °C for 3 min 30 s in 70 % deionized formamide in 2x SSC. The probe cocktail for one slide (10 μl; 50 % deionized formamide, 10 % dextran sulphate in 2× SSC) contained 50 ng of the probe and 25 μg of sonicated salmon sperm DNA (Sigma-Aldrich). Hybridization was carried out overnight. Hybridization signals of biotin-labelled probes were detected with Cy3-conjugated streptavidin (Jackson ImmunoRes. Labs. Inc., West Grove, PA, USA), followed by one round of amplification with biotinylated anti-streptavidin (Vector Laboratories, Burlingame, CA, USA) and Cy3-conjugated streptavidin. The preparations were mounted in 40 μl of DABCO antifade containing DAPI (0.5 μg/mL). The FISH experiments were performed several times on both mitotic and meiotic chromosomes.
Microscopy and image processing
FISH preparations were observed in a Zeiss Axioplan 2 epifluorescence microscope (Carl Zeiss Jena, Germany) equipped with an F-View CCD camera and AnalySIS software, version 3.2 (Soft Imaging System GmbH, Münster, Germany) or with an Olympus CCD monochrome camera XM10 and cellSens 1.9 digital imaging software (Olympus Europa Holding, Hamburg, Germany). Black-and-white images of chromosomes were recorded separately for each fluorescent dye. Images were pseudocolored (light blue for DAPI, red for Cy3) and processed with Adobe Photoshop, version 5.0.
PCR amplification of CpSAT-1 from C. pomonella gDNA
A 25 μl of the PCR reaction contained 100 ng of gDNA extracted with standard phenol–chloroform-isoamylalcohol procedure, primers (0.5 μM each), dNTPs (0.2 mM each), 2 U of Ex Taq polymerase (TaKaRa) and 1× Ex Taq buffer. The primer pair sequences were: 5′-TCTATTGAGCCCAAACACGATGG-3′ for the CpSAT-1 forward primer and 5′-TGGAGTCAGGAGTTGGTCACC-3′ for the CpSAT1-reverse primer, respectively. PCR conditions were as follow: Initial denaturation 94 °C for 3 min, 30 cycles of denaturation 94 °C for 30 s, annealing 54 °C for 30 s, extension 72 °C for 1 min, and final extension 72 °C for 2 min. The PCR products were separated on 1.5 % agarose gel in TBE, stained with ethidium bromide, documented under the UV light or used for further analyses.
RNA extraction and RT-PCR
For total RNA extraction we used both whole individuals and isolated tissues of C. pomonella. The individuals were at the following developmental stages: 1st–4th larval instars (several individuals, no sexing), 5th larval instar, pupae (both samples included one male and one female), adults (one male and one female, separately). Most tissues were isolated from 5th instar larvae (Malpighian tubules, empty gut, and heart) of both sexes in equal proportion. Testes and ovaries were taken from adults, where collection of these tissues was more feasible than at larval stages. Total RNA was isolated using RNA Blue (Top-Bio, Prague, Czech Republic) according to the manufacturer’s protocol. RNA was subsequently treated with 2 U DNase I (Life Technologies, Carlsbad, California, USA) for 15 min at 37 °C and purified. Alternatively, total RNA was extracted using RNAzol (Life Technologies) according to the manufacturer’s protocol including an optional step with 4-bromoanisole for maximal DNA removal. 5 μg of total RNA was then applied for cDNA synthesis with SuperScript III Reverse Transcriptase (Life Technologies) according to the manufacturer’s protocol. Finally, the cDNA was treated with 5 U of RNase H (TaKaRa) for 20 min at 37 °C, and 2 μl of the reaction were applied as a template for RT-PCR with CpSAT1 specific primers. The RNA extraction with subsequent RT-PCR were performed with each sample at least twice.
In order to exclude possible false positive results caused by DNA contamination, we performed two negative controls. (1) Each total RNA sample was treated with RNase A. 15 μg of total RNA were incubated with RNase A at 37 °C overnight and then 2 μl of the reaction were used in the control PCR reaction with CpSAT-1 primers. (2) We run RT-PCR with primers specific for ribosomal protein L10A (Rpl10) gene fragment including an intron. Thus, gDNA-free cDNA templates produced ca. 150 bp long fragment, while the PCR products generated from gDNA-contaminated samples would be ca. 700 bp long. The primer sequences were: 5′-TGCATGGATGCTGAGGCTTTG-3′ for Rpl10 forward primer and 5′-GAGAGCAGACCAGGGAACTTG-3′ for Rpl10 reverse primer, respectively. The PCR reaction and conditions were as described above except for the annealing temperature of Rpl10 primers, which was 58 °C.
Analysis of CpSAT-1 monomers
gDNA and cDNA PCR products were ligated into pGEM-T Easy vector (Promega) according to the manufacturer’s protocol and cloned in E. coli DH5α competent cells. The inserts were sequenced at ABI PRISM 3130xl using vector specific M13 primers. Since almost all inserts included more than one monomer of CpSAT-1, the sequences were split into individual monomers for subsequent analyses. Sequence data have been submitted to GenBank under the following accession numbers: KF421165–KF421204 for gDNA monomers and KF421205–KF421244 for cDNA monomers.
The consensus sequence was generated using default parameters where all degenerated positions were replaced manually by the most abundant nucleotide. The consensus sequence of the CpSAT-1 was applied as a query for homology search in GenBank using BLASTN search engine. The curvature propensity plot was calculated with the bend.it server (http://hydra.icgeb.trieste.it/dna/bend_it.html), using the DNase I and nucleosome positioning data (Vlahoviček et al. 2003) from CpSAT-1 consensus sequence. The curvature window size was 31 bp, the consensus sequence was applied as a dimer in order to cover its whole length.
Multiple-sequence alignments were performed using the default parameters of the program MUSCLE as implemented in the software MEGA version 5 (Tamura et al. 2011), which was also used for the phylogenetic and molecular evolutionary analyses. Pairwise distances were calculated according to the best-fit model of nucleotide evolution K2 + G (Kimura 2 parameter with gamma-distribution) selected by the BIC scores (Bayesian Information Criterion). A distance tree was built by the Neighbor-Joining (NJ) and Maximum Likelihood (ML) with pairwise-deletion option for indels and ambiguous sites. Bootstrap values were calculated based on 1000 replicates.
DNA sequence polymorphism, distribution of the polymorphic sites along the sequences, and the diversity of haplotypes were analyzed using DnaSP v.5.10.01 (Librado and Rozas 2009). The conserved and variable segments in satellite DNA sequences were defined according to Mravinac et al. (2004) by sliding window analysis using window size of 10 bp and step size of 1 bp. Windows that exhibited more than 2 standard deviations from the average variability were considered. Programs MEGA and DNAsp were also used to calculate basic parameters of genetic variability. Arlequin (Excoffier and Lischer 2010) was used to infer the connections among the haplotypes and Minimum Spaning Tree was visualized with the use of HapStar (Teacher and Griffiths 2011).
Reading frames (RFs)s and internal repeats were searched using DNA Star module GeneQuest (Lasergene, version 8.0.2.). In order to detect potential RFs over the edge of a monomer, each sequence was duplicated and tested as a dimer. Only RFs 25 aa and longer were considered, both RFs possessing or lacking the start codon were analyzed. In case of internal repeats (i.e. internal inverted and direct repeats, and dyads) the minimum sequence length was set at 8 bp. RFs and internal repeats were searched in each monomer individually. Base composition was computed for CpSAT-1 consensus sequence using the same software. Finally, cDNA monomers were searched for RNA polymerase II promoter motifs using the Neural Network Promoter Prediction Tool (http://www.fruitfly.org/seq_tools/promoter.html) (Reese 2001) from both forward and reverse strands, minimum promoter score set at 0.7.
Results
Isolation of satellite DNA
In order to study tandem repeats in the genome of C. pomonella, we digested female gDNA with 27 restriction endonucleases (REs). Out of these REs, only 14 following provided visible bands: ClaI, EcoRI, KpnI, NotI, PstI, XbaI DraI, HaeIII, HhaI, HinfI, HpaI, NspI, RsaI, and TaqI (Fermentas Canada Inc., Burlington, Canada). For further analysis we selected XbaI restriction product, where we detected a conspicuous pattern of bands consisting of a diffuse, approximately 100–200 bp long band followed by a ladder of bands ca 350, 500, 850, 1000, 1300, 1450, and 1600 bp long (Online Resource 1), which could possibly be multimers of a tandem repeat. We isolated and cloned the shortest fragment and used it as a probe for Southern hybridization with the XbaI-digested gDNA. Indeed, the XbaI restriction fragment produced a hybridization signal with a ladder-like pattern, reaching from ca. 120 bp up to several kbp, which is typical for tandem repeats (Fig. 1). Since the identified tandem repeat is the first satellite DNA in C. pomonella, we named it CpSAT-1.

Southern hybridization of CpSAT-1 probe with Cydia pomonella gDNA digested with XbaI. Note that hybridization signals form a ladder of bands corresponding to the 120 bp multiples, clearly proving that the CpSAT-1 is a tandem repeat
PCR amplification of CpSAT-1 from gDNA and cDNA
PCR with CpSAT-1 specific primers on gDNA and cDNA templates generated a ladder of bands corresponding to mono- up to octamers of the satellite sequence, with continuous smear ranging from ca. 100 bp up to several kbp (Fig. 2a). In order to detect possible CpSAT-1 transcription, we isolated total RNA from various developmental stages of C. pomonella, ranging from the 1st instar larvae to adults from both males and females. Additionally, we tested some tissues of C. pomonella, namely Malpighian tubules, gut, heart, ovaries and testes. The subsequent RT-PCR produced virtually the same pattern in all tested developmental stages and tissue samples, consisting of a smear and a ladder of bands of 120 bp multiples (Fig. 2a). To exclude possible gDNA contamination, we performed RT-PCR with primers specific for the Rpl10 gene, which amplify a gene fragment containing an intron, and therefore PCR products from cDNA and gDNA differed in their lengths. Since the gene fragment of the length typical for gDNA was missing in cDNA samples, we concluded that the obtained RT-PCR products had to originate from CpSAT1 RNA transcripts (Fig. 2b). Similarly, the PCR amplification from RNase treated samples provided no products, confirming that there was no gDNA contamination in any sample of extracted total RNA (not shown).

RT-PCR amplification of CpSAT-1 and Rpl10 from cDNA isolated from all developmental stages, both sexes, and various tissues of Cydia pomonella. a PCR amplification of CpSAT-1. Positive control (gDNA) and all samples produced a ladder of bands corresponding to CpSAT-1 multimers. b Control amplification of Rpl10 gene fragment. The first lane shows a PCR product amplified from gDNA, containing an intron. Remaining lanes show an intronless fragment only, thus proving that the RNAs used to construct cDNAs were free of gDNA contamination
Sequence analysis
In total, we analyzed 40 monomers from gDNA and 40 monomers from cDNA. The length of the monomer in the majority of sequences (95 % of cDNA and 80 % of gDNA datafile) was 120 bp, however, in a few monomers we observed short duplications. An identical 10 bp long duplication was present in 8 monomers from gDNA (in one monomer prolonged with additional 7 bp with no homology in the CpSAT-1) as well as in one monomer from cDNA. A 3-bp duplication was found in one cDNA monomer only, and was absent from gDNA monomers.
Parameters of nucleotide variability are presented in Table 1. Both groups of monomers (gDNA and RT) have very similar levels of variability. Considerably high number of variable sites (more than 1/3 of the sequence length) resulted in the formation of adequately high number of unique haplotypes but for cDNA and gDNA monomers, there was no common haplotype identified. The groups do not form any distinct clusters in the dendrograms (ESM 2); in fact, the haplotypes appear to be rather interspersed with no traceable pattern related either to its origin (gDNA or RT) or the repeats structure (momoner, dimer etc.). Similarly, in the Minimum Spanning Tree it can be seen that the haplotypes do not associate according to their origin or type (ESM 3).
Further, each monomer was searched for reading frames (RFs) coding at least 25 aa. No significant amount of monomers containing a RF starting with the start codon was found, however, RFs without start codon were abundant. Two RFs were present in the majority of monomers: RF1 covered 77.5 % of the monomer, and it was present in 67.5 % of monomers. The RF2 covered 72.5 % of the monomer, and it was present in 82 % of monomers.
Each sequence was further searched for internal repeats, i.e. dyads, inverted and direct repeats at least 8 bp long. Except for the duplication mentioned above, direct repeats were virtually absent from the CpSAT-1 sequences (1/80). In contrast, we detected 9 versions of inverted repeats and 7 types of dyads, however, most of them occurred in one or two monomers only. Nevertheless, two inverted repeats and three dyads were present in the significant portion of monomers, namely Inv1 was found in 41.25 % monomers, Inv2 in 23.75 % monomers, Dyad1a and its variant Dyad1b were present in 40 and 17.5 % monomers, respectively. Most abundant motifs and their frequencies are summarized in Table 2.
The curvature propensity plot revealed conspicuous peak reaching its magnitude 9° per helical turn around the nucleotide position 47 (Fig. 3). According to Gabrielian et al. (1997), the straight DNA motifs give values below 4–5°/helical turn, whereas curved DNA values are 9° per helical turn and higher. Therefore, we believe that this region is slightly curved.

Curvature propensity plot of CpSAT-1 dimer. The graph shows conspicuous peaks around the positions 47 and 167, regions with highest curvature propensity
Sliding window revealed two larger conservative and two variable regions of CpSAT-1, respectively. The conservative regions were located between nucleotide positions 33–48 and 64–128 bp (including 10-bp duplicated region), whereas the variable regions were found between nucleotides 7–21 and 49–57 (Fig. 4).

Sliding window analysis of CpSAT-1 sequence variability. Identification of conserved (shaded) parts in the CpSAT1 monomers using sliding window of 10 bp. Average diversity (Pi) is indicated with dashed line, while the average diversity ±2 SD is indicated with dotted line. Comparison of 80 CpSAT-1 monomers revealed local conservative regions between positions 33–48 and 64–128, respectively. Black bars represent insertions located in some monomers. First insertion (72nd–81st bp) was present in 8 gDNA and 1 cDNA monomer, respectively, second insertion (87th–93rd bp) was present in one gDNA monomer, and the third insertion (112th–114th bp) was present in one cDNA monomer only
The nucleotide composition of CpSAT-1 consensus sequence was as follows: T 26.7 %, C 21.7 %, A 26.7 %, and G 25.0 %. Thus, the CpSAT-1 is slightly AT rich. The graphic overview of consensus sequence GC content in Fig. 3 shows that the distribution of GC nucleotides throughout the CpSAT-1 sequence is not uniform and reaches its maximum at both ends of the monomer. Further, we attempted to predict the RNA polymerase II promoter in the cDNA monomers using the Neural Network Promoter Prediction Tool. Nevertheless, we failed to find any promoter motif within the CpSAT-1 sequence.
Finally, we searched GenBank for CpSAT-1 consensus sequence homologs and found a single sequence with a high similarity (93 %, E value = 3e–35). Interestingly, the sequence was annotated as microsatellite sequence from C. pomonella, clone CP5.173 (Accession number DQ394030.1), and contained one monomer of CpSAT-1. Screening for CpSAT-1 consensus sequence homolog in RepBase (http://www.girinst.org/repbase) did not bring any results (Kohany et al. 2006).
FISH analysis of chromosomal distribution of CpSAT-1
Localization of sequences in eukaryotic organisms is usually performed on mitotic chromosomes. Since lepidopteran chromosomes are generally very small, we used both the chromosomes at mitotic metaphase and meiotic prophase I (pachytene bivalents) to get better resolution.
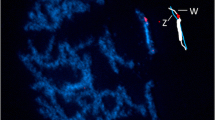
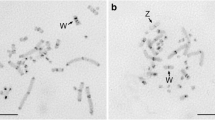
FISH showed that CpSAT-1 is present virtually in every chromosome, although its abundance differs among individual chromosomes ranging from very abundant in some chromosomes to nearly missing in the others (Fig. 5). It is noteworthy that the satellite is not abundant on the W chromosome, the only large heterochromatin compartment of the C. pomonella genome. We found several clusters of hybridization signals scattered along the W chromosome. In comparison with the W chromosome, the Z chromosome carries more CpSAT-1 clusters concentrated mainly in the internal part. There are some clusters in the W and Z chromosomes, which seem to co-localize.

Fluorescence in situ hybridization of CpSAT-1 probe on mitotic metaphase (a), and pachytene (b, d, f, and h) chromosomes of Cydia pomonella female. c, e, and g show schematic drawings of the WZ bivalents (W white; Z blue) with presumed W (red) and Z (green) CpSAT-1 clusters according to individual WZ bivalents seen in (d, f, and h), respectively. A similar WZ scheme is shown in the lower left corner of (b). Hybridization signals of the CpSAT-1 probe (red) are present on all chromosomes, but their intensity differs among the chromosomes, showing various abundance of the CpSAT-1. Interestingly, the W chromosome contains a low amount of the CpSAT-1, whereas the Z chromosome is relatively CpSAT-1 rich. Chromosomes were counterstained with DAPI (blue). Bar = 10 μm
Discussion
Finished and ongoing whole-genome sequencing projects in Lepidoptera are mainly focused on protein-coding genes, while analyses of repetitive sequences are often absent or limited to the mobile elements (Mita et al. 2004; Xia et al. 2004; The International Silkworm Genome Consortium 2008). So far, there has been only three lepidopteran satellite sequences described (Lu et al. 1994; Mandrioli et al. 2003; Mahendran et al. 2006), which is in contrast with the increasing number of evidence about satDNA significance for the genome. In this work, we present a new satDNA for Lepidoptera and the first found in the codling moth, C. pomonella.
CpSAT-1 and functional motifs
We tested the CpSAT-1 for transcriptional activity in all instars, both sexes and several tissues and proved CpSAT-1 transcripts in all samples. This could mean that CpSAT-1 has a function in the genome. In order to uncover potential function of the satellite, we searched for some specific structural features, such as ORFs, internal repeats, and promoters. We found abundant reading frames covering most of the monomer length. However, they lack the start codon indicating that the CpSAT-1 is not translated. Some inverted repeats and dyads occurred in a high number of monomers (see Table 2). Interestingly, none of the internal repeats was located in the conservative region (except for a half of the Inv1); instead, the repeats concentrated in the neighbouring variable region. Similarly, the segment with the highest curvature propensity only partly co-localized with the conservative region. Nevertheless, our test of ten randomly selected monomers showed that this region remains curved in the majority of them (9/10) regardless of minor sequence differences. These findings suggested that either the internal repeats have no function in the CpSAT-1 or the hypothetical function is performed by a subset of monomers with intact IRs, maintained by selection. Such feature was described in the human centromeric alpha-satellite DNA, where proper centromere function is maintained by a limited number of monomers (reviewed by Alexandrov et al. 2001). However, sequence analysis of CpSAT-1 monomers obtained from cDNA and gDNA showed high variability among the monomers which was comparable within the cDNA and gDNA group (36 for gDNA and 31 in RT out of 40 samples, no haplotype is shared between the groups). Therefore, the transcribed monomers probably do not represent a conservative subset of the CpSAT-1, which would be expected if the CpSAT-1 has a function. Finally, we failed to find any RNA polymerase II promoter in the CpSAT-1 sequence. This suggests that either the CpSAT-1 transcription is performed by other RNA polymerase, as proposed for Schistosoma mansoni Smα satDNA (Ferbeyre et al. 1998), or the promoter site is located in flanking regions or sequences interspersed in CpSAT-1 arrays. The transcription of the satellite could be also driven by regulation sequences of an adjacent gene or genes and represents just a transcriptional noise without any function. Considering the scattered distribution and quite high abundance of the CpSAT-1 in the genome, the presence of such regulation sequences in the neighbourhood is highly probable.
CpSAT-1 location in the genome of Cydia pomonella
Moths and butterflies (Lepidoptera) have a female heterogametic sex chromosome system, where most females have a pair of WZ sex chromosomes and males have two Z chromosomes. While the Z chromosome is gene rich and mostly consists of euchromatin, thus resembling the autosomes, the W is usually made up of heterochromatin and mostly carries repetitive sequences (Abe et al. 2005; Fuková et al. 2007; Abe et al. 2010; Traut et al. 2013), but only few or no genes (reviewed in Sahara et al. 2012). Furthermore, it was shown that the Z chromosome gene content is highly conserved in Lepidoptera (Van’t Hof et al. 2012). In contrast, the W chromosomes differ greatly even among related species (e.g. Vítková et al. 2007).
We localized CpSAT-1 on both mitotic and meiotic chromosomes of female codling moth by means of FISH. Hybridization signals of the CpSAT-1 probe were present in all chromosomes, resembling the location of the AmTFR satDNA from Antheraea myllita (Mahendran et al. 2006), however, distribution and intensity of signals differed greatly among individual chromosomes. More specifically, the CpSAT-1 was found to be abundant on some chromosomes, but few in number on other chromosomes. Since satDNAs are known to be preferentially located in heterochromatin areas, it is surprising that the CpSAT-1 is underrepresented on the W chromosome, the largest heterochromatin element in the codling moth genome, which is mainly composed of ubiquitous repetitive DNA sequences (Fuková et al. 2007). However, the satellite is relatively frequent on the Z chromosome. This difference may be caused either by loss of CpSAT-1 sequences in the non-recombining W chromosome during its genetic erosion (e.g. Abe et al. 2005; Vítková et al. 2007), or the CpSAT-1 could spread in the Z chromosome after the suppression of recombination between these originally homologous sex chromosomes. Further, a significant part of the codling moth Z chromosome originates from a fusion of the Z with an autosome (Nguyen et al. 2013). Therefore, the Z chromosome located CpSAT-1 sequences could originate and possibly spread from this formerly autosomal part. However, as suggested by Šíchová et al. (2013), not only the Z chromosome but possibly also the tortricid W chromosome had originated by fusion between an ancestral W chromosome and an autosome. Finally, considering the relatively high abundance of CpSAT-1 on the autosomes, and a high turnover of the sequences on the W chromosome, which does not concern only Lepidoptera (Abe et al. 2005; Vítková et al. 2007; Traut et al. 2013) but is a general feature of the W and Y chromosomes in other organisms (Charlesworth and Charlesworth 2000), we propose the hypothesis of CpSAT-1 decay during degeneration of the W chromosome. However, ruling out any of the hypotheses would require further investigation of either C. pomonella W chromosome sequence or the abundance and distribution of CpSAT-1 in related species.
Taken together, the absence of promoter sequence, omnipresent transcription in all tested tissues and developmental stages, high sequence variability comparable in both the gDNA and cDNA derived monomers, the absence of potentially functional motifs in conservative regions, and uneven distribution in the genome support the hypothesis that the CpSAT-1 has no function in the genome. However, this conclusion requires further research.
References
Abe H, Mita K, Yasukochi Y et al (2005) Retrotransposable elements on the W chromosome of the silkworm, Bombyx mori. Cytogenet Genome Res 110:144–151. doi:10.1159/000084946
Abe H, Fujii T, Shimada T, Mita K (2010) Novel non-autonomous transposable elements on W chromosome of the silkworm, Bombyx mori. J Genet 89:375–387
Alexandrov I, Kazakov A, Tumeneva I et al (2001) Alpha-satellite DNA of primates: old and new families. Chromosoma 110:253–266. doi:10.1007/s004120100146
Ausubel FM, Brent R, Kingston RE et al (2003) Current protocols in molecular biology. Wiley, New York
Bardella VB, da Rosa JA, Vanzela ALL (2014) Origin and distribution of AT-rich repetitive DNA families in Triatoma infestans (Heteroptera). Infect Genet Evol 23:106–114. doi:10.1016/j.meegid.2014.01.035
Biessmann H, Donath J, Walter MF (1996) Molecular characterization of the Anopheles gambiae 2L telomeric region via an integrated transgene. Insect Mol Biol 5:11–20. doi:10.1111/j.1365-2583.1996.tb00035.x
Bizzaro D, Manicardi GC, Bianchi U (1996) Chromosomal localization of a highly repeated EcoRI DNA fragment in Megoura viciae (Homoptera, Aphididae) by nick translation and fluorescence in situ hybridization. Chromosome Res 4:392–396. doi:10.1007/BF02257275
Charlesworth B, Charlesworth D (2000) The degeneration of Y chromosomes. Philos Trans R Soc Lond B Biol Sci 355:1563–1572. doi:10.1098/rstb.2000.0717
Charlesworth B, Sniegowski P, Stephan W (1994) The evolutionary dynamics of repetitive DNA in eukaryotes. Nature 371:215–220. doi:10.1038/371215a0
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Res 10:564–567. doi:10.1111/j.1755-0998.2010.02847.x
Feliciello I, Akrap I, Ugarković Đ (2015) Satellite DNA modulates gene expression in the beetle Tribolium castaneum after heat stress. PLoS Genet 11:e1005466. doi:10.1371/journal.pgen.1005466
Ferbeyre G, Smith J, Cedergren R (1998) Schistosome satellite DNA encodes active hammerhead ribozymes. Mol Cell Biol 18:3880–3888
Fuková I, Nguyen P, Marec F (2005) Codling moth cytogenetics: karyotype, chromosomal location of rDNA, and molecular differentiation of sex chromosomes. Genome 1092:1083–1092. doi:10.1139/G05-063
Fuková I, Traut W, Vítková M et al (2007) Probing the W chromosome of the codling moth, Cydia pomonella, with sequences from microdissected sex chromatin. Chromosoma 116:135–145. doi:10.1007/s00412-006-0086-0
Gabrielian A, Vlahovicek K, Pongor S (1997) Distribution of sequence-dependent curvature in genomic DNA sequences. FEBS Lett 406:69–74. doi:10.1016/S0014-5793(97)00236-6
Heckmann S, MacAs J, Kumke K et al (2013) The holocentric species Luzula elegans shows interplay between centromere and large-scale genome organization. Plant J 73:555–565. doi:10.1111/tpj.12054
Kohany O, Gentles AJ, Hankus L, Jurka J (2006) Annotation, submission and screening of repetitive elements in Repbase: repbaseSubmitter and Censor. BMC Bioinform 7:474. doi:10.1186/1471-2105-7-474
Li Y-X, Kirby ML (2003) Coordinated and conserved expression of alphoid repeat and alphoid repeat-tagged coding sequences. Dev Dyn 228:72–81. doi:10.1002/dvdy.10355
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452. doi:10.1093/bioinformatics/btp187
Lorite P, Renault S, Bigot S et al (2002) Genomic organization and transcription of satellite DNA in the ant Aphaenogaster subterranea (Hymenoptera, Formicidae). Genome 616:609–616. doi:10.1139/G02-022
Lu YJ, Kochert GD, Isenhour DJ, Adang MJ (1994) Molecular characterization of a strain-specific repeated DNA sequence in the fall armyworm Spodoptera frugiperda (Lepidoptera: Noctuidae). Insect Mol Biol 3:123–130. doi:10.1111/j.1365-2583.1994.tb00159.x
Mahendran B, Acharya C, Dash R et al (2006) Repetitive DNA in tropical tasar silkworm Antheraea mylitta. Gene 370:51–57. doi:10.1016/j.gene.2005.11.010
Mandrioli M, Bizzaro D, Manicardi GC et al (1999) Cytogenetic and molecular characterization of a highly repeated DNA sequence in the peach potato aphid Myzus persicae. Chromosoma 108:436–442. doi:10.1007/s004120050395
Mandrioli M, Manicardi GC, Marec F (2003) Cytogenetic and molecular characterization of the MBSAT1 satellite DNA in holokinetic chromosomes of the cabbage moth, Mamestra brassicae (Lepidoptera). Chromosome Res 11:51–56. doi:10.1023/A:1022058032217
Masumoto H, Masukata H, Muro Y et al (1989) A human centromere antigen (CENP-B) interacts with a short specific sequence in alphoid DNA, a human centromeric satellite. J Cell Biol 109:1963–1973. doi:10.1083/jcb.109.5.1963
Metz A, Soret J, Vourc’h C et al (2004) A key role for stress-induced satellite III transcripts in the relocalization of splicing factors into nuclear stress granules. J Cell Sci 117:4551–4558. doi:10.1242/jcs.01329
Mita K, Kasahara M, Sasaki S et al (2004) The genome sequence of silkworm, Bombyx mori. DNA Res 11:27–35. doi:10.1093/dnares/11.1.27
Mravinac B, Plohl M, Ugarković D (2004) Conserved patterns in the evolution of Tribolium satellite DNAs. Gene 332:169–177. doi:10.1016/j.gene.2004.02.055
Nguyen P, Sýkorová M, Šíchová J et al (2013) Neo-sex chromosomes and adaptive potential in tortricid pests. Proc Natl Acad Sci USA 110:6931–6936. doi:10.1073/pnas.1220372110
Palomeque T, Lorite P (2008) Satellite DNA in insects: a review. Heredity (Edinb) 100:564–573. doi:10.1038/hdy.2008.24
Plohl M, Luchetti A, Mestrović N, Mantovani B (2008) Satellite DNAs between selfishness and functionality: structure, genomics and evolution of tandem repeats in centromeric (hetero)chromatin. Gene 409:72–82. doi:10.1016/j.gene.2007.11.013
Reese MG (2001) Application of a time-delay neural network to promoter annotation in the Drosophila melanogaster genome. Comput Chem 26:51–56. doi:10.1016/S0097-8485(01)00099-7
Renault S, Rouleux-Bonnin F, Periquet G, Bigot Y (1999) Satellite DNA transcription in Diadromus pulchellus (Hymenoptera). Insect Biochem Mol Biol 29:103–111. doi:10.1016/S0965-1748(98)00113-1
Rouleux-Bonnin F, Renault S, Bigot Y, Periquet G (1996) Transcription of four satellite DNA subfamilies in Diprion pini (Hymenoptera, Symphyta, Diprionidae). Eur J Biochem 238:752–759. doi:10.1111/j.1432-1033.1996.0752w.x
Rouleux-Bonnin F, Bigot S, Bigot Y (2004) Structural and transcriptional features of Bombus terrestris satellite DNA and their potential involvement in the differentiation process. Genome 47:877–888. doi:10.1139/g04-053
Sahara K, Marec F, Traut W (1999) TTAGG telomeric repeats in chromosomes of some insects and other arthropods. Chromosome Res 7:449–460. doi:10.1023/A:1009297729547
Sahara K, Yoshido A, Traut W (2012) Sex chromosome evolution in moths and butterflies. Chromosome Res 20:83–94. doi:10.1007/s10577-011-9262-z
Saiga H, Edström JE (1985) Long tandem arrays of complex repeat units in Chironomus telomeres. EMBO J 4:799–804
Šíchová J, Nguyen P, Dalíková M, Marec F (2013) Chromosomal evolution in tortricid moths: conserved karyotypes with diverged features. PLoS One 8:e64520. doi:10.1371/journal.pone.0064520
Spence JM, Blackman RL, Testa JM, Ready PD (1998) A 169-base pair tandem repeat DNA marker for subtelomeric heterochromatin and chromosomal rearrangements in aphids of the Myzus persicae group. Chromosome Res 6:167–175. doi:10.1023/A:1009251415941
Tamura K, Peterson D, Peterson N et al (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. doi:10.1093/molbev/msr121
Teacher AGF, Griffiths DJ (2011) HapStar: automated haplotype network layout and visualization. Mol Ecol Resour 11:151–153. doi:10.1111/j.1755-0998.2010.02890.x
The International Silkworm Genome Consortium (2008) The genome of a lepidopteran model insect, the silkworm Bombyx mori. Insect Biochem Mol Biol 38:1036–1045. doi:10.1016/j.ibmb.2008.11.004
Traut W, Szczepanowski M, Vítková M et al (2007) The telomere repeat motif of basal Metazoa. Chromosome Res 15:371–382. doi:10.1007/s10577-007-1132-3
Traut W, Vogel H, Glöckner G et al (2013) High-throughput sequencing of a single chromosome: a moth W chromosome. Chromosome Res 21:491–505. doi:10.1007/s10577-013-9376-6
Van’t Hof A, Nguyen P, Dalíková M et al (2012) Linkage map of the peppered moth, Biston betularia (Lepidoptera, Geometridae): a model of industrial melanism. Heredity 110:283–295. doi:10.1038/hdy.2012.84
Vítková M, Fuková I, Kubíčková S, Marec F (2007) Molecular divergence of the W chromosomes in pyralid moths (Lepidoptera). Chromosome Res 15:917–930. doi:10.1007/s10577-007-1173-7
Vlahoviček K, Kaján L, Pongor S (2003) DNA analysis servers: plot.it, bend.it, model.it and IS. Nucl Acids Res 31:3686–3687. doi:10.1093/nar/gkg559
Volpe TA, Kidner C, Hall IM et al (2002) Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science 297:1833–1837. doi:10.1126/science.1074973
Xia Q, Zhou Z, Lu C et al (2004) A draft sequence for the genome of the domesticated silkworm (Bombyx mori). Science 306:1937–1940. doi:10.1126/science.1102210
Acknowledgments
We thank Marie Korchová for rearing of the codling moth and František Marec for critical reading of the manuscript. This research was funded by Grants KJB501410901 of the Grant Agency of the Academy of Sciences of the Czech Republic and 523/09/2106 of the Grant Agency of the Czech Republic (GACR). MD and MS were supported from GACR Grant 14-22765S. Additional support from grant 052/2013/P of the Grant Agency of the University of South Bohemia is also acknowledged. Finally, we wish to thank Juan Pedro M. Camacho, Francisco J. Ruiz-Ruano (both University of Granada, Spain), and two anonymous reviewers for valuable comments to the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Věchtová, P., Dalíková, M., Sýkorová, M. et al. CpSAT-1, a transcribed satellite sequence from the codling moth, Cydia pomonella . Genetica 144, 385–395 (2016). https://doi.org/10.1007/s10709-016-9907-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10709-016-9907-0