
Abstract
In order to develop a rice population with improved important traits such as flowering time, we developed 2,911 M2 targeting-induced local lesions in genomes (TILLING) lines by irradiating rice seeds with γ-rays. In all, 15 M3 lines were obtained from 3 different M2 lines that exhibited an early-maturing phenotype: these plants matured approximately 25 days faster than wild-type (WT) plants. To identify genome-wide DNA polymorphisms, we performed whole-genome resequencing of both the plant types, i.e., WT and early-maturing TILLING 1 (EMT1), and obtained mapped reads of 118,488,245 bp (99.53 %) and 128,489,860 bp (99.72 %), respectively; Nipponbare was used as the reference genome. We obtained 63,648 and 147,728 single nucleotide polymorphisms (SNPs) and 33,474 and 31,082 insertions and deletions (InDels) for the WT and EMT1, respectively. Interestingly, there was a higher number of SNPs (2.6-fold) and slightly lower number of InDels (0.9-fold) in EMT1 than in WT. The expression of at least 202 structurally altered genes was changed in EMT1, and functional enrichment analysis of these genes revealed that their molecular functions were related to flower development. These results might provide a critical insight into the regulatory pathways of rice flowering.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
More than 2,700 cultivars obtained either as direct mutants or derived from 170 different plant species have been released worldwide in 60 countries (http://mvgs.iaea.org). Mutation induction continues to contribute to crop improvement via the creation of new alleles by using physical mutagens such as fast neutrons, X-rays, and γ-rays and chemical mutagens such as sodium azide and ethyl methanesulfonate. Recently, novel physical mutagen sources such as ion beam radiation and cosmic rays have been suggested to be effective for creating mutations. Therefore, mutation techniques are generally believed to be useful in creating novel traits and enhancing genetic diversity. Variations in quantitative traits, for example, high levels of lysine and essential amino acids in rice, were created via γ-ray irradiation for the breeding of crops with mutations (Kim et al. 2004a, b).
The detection of DNA polymorphisms for genetic variation has been an integral part of plant breeding through marker-assisted selection and is useful for germplasm collections. Many molecular markers have been developed rapidly for genetic mapping and identifying novel genes (Jones et al. 2009); however, the available molecular marker systems are somewhat limited for the genome-wide detection of genetic polymorphisms. Recently, the application of next-generation sequencing technologies has allowed the discovery of large numbers of genome-wide DNA polymorphisms. In rice, Li et al. (2012) reported genome-wide variations among 3 elite restorer lines compared with the sequence of indica cultivar 93–11 by using Solexa sequencing technology. In addition, more than 2.8 million genome-wide polymorphisms were discovered among 6 elite indica rice inbreds by using whole-genome sequencing (Subbaiyan et al. 2012). These genome-wide levels of DNA polymorphisms represent useful and cost-effective resources for molecular breeding.
Flowering time is an important agronomic trait for crop improvement under various environmental conditions. This process is generally intricately regulated by a complicated network of signaling pathways. A comparative genomics approach suggested an ancient core photoperiod has diverged into lineage-specific pathways during evolution in plants (Higgins et al. 2010). For example, the circadian clock, photoperiod, autonomous and gibberellin pathways, and response to temperature play important roles in flowering time control across plants, including monocotyledons and tropical and temperate dicotyledons (Higgins et al. 2010; Mouradov et al. 2002). In rice, many flowering time-related genes have been identified, e.g., Hd1 (heading date 1), Ehd1 (early heading date 1), and Ghd7 (grain number, plant height, and heading date 7; Yano et al. 2000; Doi et al. 2004; Xue et al. 2008). However, genome-wide analysis of flowering time has not yet been performed.
Recently, the targeting-induced local lesions in genomes (TILLING) methodology, which is an excellent reverse genetics technique, has been reported to facilitate the functional identification of mutations in specific genes in functional genomics research (Till et al. 2003). Previously, we developed a TILLING rice population via the application of γ-ray irradiation to a rice variety (cv. Donganbyeo), followed by the evaluation of its genetic diversity (Cho et al. 2010). The Donganbyeo variety was developed from a cross between Milyang 95 and HR5119-12-1-5 line at the National Honam Agricultural Experiment Station, RDA, Korea (Shin et al. 1997). This cultivar exhibits a mid- or late-flowering time with a growth duration of approximately 122 days from transplanting to harvesting and high adaptability for direct-seeded cultivation with lodging resistance (Shin et al. 1997).
In the present study, we selected three independent early-flowering time M2 TILLING lines from the 2,911 M2 TILLING lines and subsequently developed M5 TILLING lines. Whole-genome resequencing of an early-flower time TILLING line as well as Donganbyeo was performed to evaluate the genetic changes in these plants. In addition, differences in expression patterns between both the plants were evaluated using genome-wide transcriptome analysis. Interestingly, one gene encoding a leucine-rich repeat receptor-like kinase (LRR-RLK) showed a changed function in the TILLING line.
Materials and methods
Plant materials
A total of 2,911 M2 TILLING lines were developed after 200 and 300 Gy γ-ray irradiation was applied to Donganbyeo (wild-type, WT) at the Korea Atomic Energy Research Institute, Korea, as described elsewhere (Cho et al. 2010). The seedlings of the TILLING lines were grown in a greenhouse and then transferred to the Kangwon National University Farm (latitude, 37.52N; longitude, 127.53E; Chuncheon, Korea). Approximately, 15,000 M3 and M4 TILLING lines were developed through self-pollination of the M2 lines. The heading dates at which panicles emerged from the flag leaf of the M3 to M5 TILLING lines were surveyed in the field over 3 years (2010–2012) and comparison to those of the WT plants.
Fingerprinting assay
Genomic DNA was extracted from the plants by using the modified cetyltrimethylammonium bromide (CTAB) method (Saghai-Maroof et al. 1984). Amplified fragment length polymorphism (AFLP) fingerprinting was conducted as described by Vos et al. (1995) with some modifications. The fragments were visualized using the SILVER SEQUENCE™ DNA Sequencing System (Promega, USA) according to the manufacturer’s instructions. The polymorphic fragments were isolated and characterized as described previously (Cho et al. 2010). The full-length sequence and 1-kb upstream region of a gene of interest were isolated using a Genome Walker Universal Kit (Clontech, USA) according to the manufacturer’s instructions. Gene expression was evaluated using semi-quantitative real time-polymerase chain reaction (RT-PCR) as described by Lim et al. (2010). The list of gene-specific primers is shown in Supplementary Table 1.
Read mapping
Genomic DNA from the WT and M5 early-maturing TILLING (EMT) lines was extracted from the leaves by using the CTAB method (Saghai-Maroof et al. 1984) and was used to construct a whole-genome resequencing library by using an Illumina HiSeq2000 according to the manufacturer’s instructions.
Quality control of the trimmed short reads of pair-end sequencing was conducted using the next-generation sequencing-quality control (NGS QC) toolkit with a PHRED quality score of 30 (Patel and Jain 2012). The trimmed reads were aligned to the Nipponbare reference genome sequence by using the Burrows–Wheeler aligner (Li and Durbin 2010). The complete genome sequence of Nipponbare was downloaded from Phytozome v8.0 (http://www.phytozome.net/). SAMtools was used to convert the binary format (BAM) of the generated sequence alignment map (SAM; Li et al. 2009). The potential PCR duplicates were removed for more accurate variant detection by trimming the BAM files by using Picard software (http://picard.sourceforge.net/). The trimmed BAM files were sorted using Picard and then single nucleotide polymorphisms (SNPs) and insertions/deletions (InDels) were detected using SAMtools (Li et al. 2009). Variant call format files of WT and EMT1 were annotated according to their chromosomal positions on Phytozome v8.0 by using SNP Effect Predictor (http://snpeff.sourceforge.net/). A comparative analysis between WT and EMT1 was conducted using Vcftools (http://vcftools.sourceforge.net/docs.html). The assembled whole-genome sequences of WT were generated using SAMtools and were then used to align the short reads from EMT1 for the detection of structural variations (SVs) and unmapped regions. The SVs of EMT1 were compared with the assembled genome sequence of WT and then detected using GASVpro-HQ (Sindi et al. 2012). The unmapped genes were detected by sorting the unmapped short reads using SAMtools on the aligned BAM files and then dividing the reads of paired ends by using Hydra 0.5.3 (http://www.mybiosoftware.com/sequence-analysis/4020). The divided short reads were aligned using the SOAPdenovo tool with the default parameters (http://soap.genomics.org.cn/soapdenovo.html). The assembled consensus sequence of unmapped short reads was annotated using a BLASTn search with a cutoff value of e−20 and compared with the Nipponbare reference genome.
Microarray hybridization
Leaf and panicle tissues were harvested from the young panicle stage of WT and EMT1. Panicles were ≤3 cm in length. Total RNA was extracted using the TRIzol® reagent (Invitrogen, USA), and microarray hybridization was performed using the Affymetrix GeneChip® Rice Genome Array (Affymetrix, USA). The microarray data of leaves and panicles from both the plants were normalized using the reduced major axis (RMA) method and the MAS5 detection call. Functional enrichment analysis of co-expressed genes was conducted using AgriGO tools (http://bioinfo.cau.edu.cn/agriGO/) with default parameters, and then the expression heatmaps were visualized using MEV v4.6 (http://www.tm4.org/).
Subcellular localization
A mutated gene (Os11g40890) from EMT1 was amplified using BamHI-F1 (5′-GAGAGGATCCATGCCACAGCAGCATCTTC-3′) and KpnI-R1 (5′-GAGAGGTACCGTTGATCAGTGCATGATATTT-3′) primers. The PCR product was cleaved using BamHI and KpnI and then inserted into the pBIN-35S expression vector. The recombinant vector was transformed into Agrobacterium and then incubated in yeast extract broth (YEB) medium with selection antibiotics at 28 °C for 16 h and centrifuged at 200 rpm. After centrifugation, the pelleted cells were resuspended in 5 mL infiltration buffer (10 mM MES, 10 mM MgCl2, pH 5.6) and then rinsed three times with fresh buffer. The diluted culture was co-infiltrated with the p19 silencing suppressor construct into Nicotiana benthamiana leaves as described elsewhere (Li et al. 2002). The expression of 35S::mOs11g40890-EGFP was monitored at 3 days after infiltration. Fluorescent images were captured using a multi-photon confocal laser scanning microscope (LSM 510 META NLO; Carl Zeiss).
In vitro kinase assay
For the in vitro kinase assay, a mutated gene (mOs11g40890) was amplified using NdeI-F1 (5′-GAGACATATGATGAATTCCCTCACTGGTGA-3′) and NotI-R1 (5′-GAGAGCGGCCGCCTCAGTGATTGATCAGTGCATG-3′) primers. The PCR product was cleaved with NdeI and NotI and then inserted into the pMAL-c5x expression vector (New England Biolabs, Beverly, MA, USA). The recombinant was expressed in Escherichia coli strain BL21 (DE3) pLysS as a fusion protein with maltose-binding protein. The expressed protein was purified using amylose resin via affinity chromatography (New England Biolabs, Beverly, MA, USA), and the purified protein was quantified using the Bradford assay. The purified protein was incubated with kinase assay sample buffer (1 mM dithiothreitol (DTT), 50 mM 4-(2-hydroxyethyl)-1-piperazineëthanesulfonic acid (HEPES), 10 mM MnCl2, 10 mM MgCl2) and [γ-32P]ATP for 20 min at 25 °C and transferred to an incubator at 70 °C for 30 min. The samples were electrophoresed on a 10 % acrylamide gel for approximately 1 h at 200 V. The gel was stained using Coomassie brilliant blue staining buffer for 30 min, transferred to a destaining buffer, and then to a fixing buffer. The fixed gel was transferred to a drying machine with a vacuum and dried for 1 h at 80 °C. The gel was transferred to a cassette and exposed to a film for 30 min in a deep freezer. The Arabidopsis BRI1 protein was used as a positive control (Li et al. 2002).
Results
Phenotypes and genotypes of the EMT lines
Five seeds from each 2,911 rice M2 TILLING line exposed to γ-radiation were grown in a paddy field, resulting in the cultivation of plants from three lines (hereafter, named early-maturing TILLING lines 1, 2, and 3 and designated as EMT1, 2, and 3); these lines exhibited an early-maturing phenotype (approximately 25 days) compared to the WT plants. Subsequently, M5 TILLING lines were developed, and all progeny from EMT1, EMT2, and EMT3 showed uniformly earlier heading dates than those of the WT plants. For example, when M5 EMTs and WT plants were transferred to the paddy field on May 18, 2012, their heading dates were July 15 and August 9, 2012, respectively.
A survey of the genetic diversity between the WT and M3 EMT plants was conducted using a fingerprinting method, i.e., AFLP analysis, with 64 primer combinations (Supplementary Table 1). The polymorphic fragments of the three EMTs showed the same pattern, but they were different from the patterns of WT plants. The polymorphic fragments were divided into two groups: 30 EMT-specific and 36 WT-specific polymorphic fragments (Supplementary Table 2). The genomic changes in the EMTs were investigated by obtaining the sequences of 15 randomly selected polymorphic fragments (5 WT-specific and 10 EMT-specific) and then matching them to the rice genome database by using BLASTn (Table 1). Interestingly, a fragment amplified by the E6M6 primer was matched to the exon region of one rice gene model, Os11g40890, and a comparison between the sequences of the EMTs and the Nipponbare reference sequence showed a difference in an EcoRI site (Supplementary Fig. 1). E6M6 fragment sequences showed that the same sequence was altered in EMT1, 2, and 3 (Supplementary Fig. 1).
The phenotypic differences between the M5 EMT1 and WT plants were evaluated. Clear differences were observed between both plant types for plant height, panicle length, grain number and effective grains per panicle, seed setting rate, weight per 1,000 grains, length and number of internodes, and heading date (Table 2). Heading date was earlier in EMT1 (102.4 days ± 1.4) than in WT (126.2 days ± 0.9). Further, EMT1 plants were taller (84.3 cm ± 4.6) than WT plants (88.4 cm ± 4.9). For agronomic traits related to yield potential, panicle length was longer in EMT1 (19.3 cm ± 1.4) than in WT (17.12 cm ± 1.37). The number of grains (151.5 ± 17.5) and effective grains per panicle (144.9 ± 18.2) of EMT1 were higher than those of WT (73.6 ± 9.9 and 64.2 ± 10.8, respectively), resulting in a significant difference in the seed setting rate percentage between EMT1 (86.9 % ± 5.6) and WT (95.6 % ± 1.7). In contrast, weight per 1,000 grains was greater in WT (21.5 g ± 0.3) than in EMT1 (20.9 g ± 0.4).
Whole genome resequencing
The genome-wide variation between WT and EMT1 plants was evaluated by constructing two DNA libraries for both plant types and then sequencing them (Table 3). A total of 119,048,626 bp and 128,846,054 bp were generated for WT and EMT1, respectively. The high-quality raw datasets were aligned with the reference genome sequence of Nipponbare, resulting in ~20× coverage by using several filtering criteria for redundant reads. The consensus sequence of WT and EMT1 covered approximately 99.53 and 99.72 % of the reference genome, respectively, indicating that the generated dataset was highly relevant. In contrast, heterozygous reads were approximately 0.28 and 0.16 % for WT and EMT1, respectively.
Genome-wide polymorphisms between WT and EMT1 plants were identified by detecting SNPs and InDels by using SAMtools for variant calling on the basis of comparisons with the Nipponbare reference sequence. In all, 63,648 and 147,728 SNPs and 33,474 and 31,082 InDels were obtained for WT and EMT1, respectively, with 10,907 SNPs and 8,483 InDels in common (Fig. 1a, b). Interestingly, there was a higher number of SNPs (2.6-fold) and slightly lower number of InDels (0.9-fold) in EMT1 than in WT. The genetic relationship between the samples and the reference genome sequence was determined by constructing a phylogenetic tree by using authentic SNPs and InDels (Fig. 1c), which revealed a closer relationship between WT and Nipponbare than between WT and EMT1. This finding might be attributed to the remarkable change in the EMT1 genome following γ-ray irradiation.

Variations between WT and EMT1 and phylogenetic tree analysis. a, b Dendrograms of DNA polymorphisms of SNPs and InDels, respectively. c Phylogenetic tree constructed using neighbor joining method with authentic collected sequences of SNPs
The frequencies of SNPs and InDels on individual chromosomes of WT and EMT1 were surveyed by comparing with the reference sequence (Table 4). SNPs and InDels were divided into two groups, i.e., common and uncommon polymorphisms, which indicated the polymorphic occurrences on the same chromosomal locations in WT and/or EMT1 by comparing with the Nipponbare genome sequences. A broad range of change rates of the common SNPs and InDels was found among individual chromosomes, resulting in ranges of 1/14,891 bp (chr. 8) to 1/233,839 bp (chr. 7) and 1/18,021 bp (chr. 4) to 1/179,985 bp (chr. 7), respectively. For the uncommon polymorphisms of WT, the change rates of SNPs exhibited the highest value of 1/1,424 bp for chromosome 12 and the lowest value of 1/35,336 bp for chromosome 2, while those of InDels were 1/3,741 bp for chromosome 12 and 1/74,097 bp for chromosome 2. In contrast, both chromosomes 6 and 11 of EMT1 exhibited the highest change rates of SNPs and InDels.
The authentic SNPs were classified into two groups, i.e., transitions (C/T and G/A) and transversions (C/G, T/A, A/C, and G/T), on the basis of nucleotide substitutions. For both the common and uncommon SNPs, the transition/transversion ratios exhibited a range of 2.23–2.53, indicating that transitions occurred at a higher frequency than transversions (Table 5). Interestingly, the frequency of transitions and transversions in EMT1 was considerably higher than that in WT, supporting the hypothesis that high levels of DNA alterations occurred in the EMT1 genome following γ-ray irradiation.
The functional roles of the SNPs and InDels were further understood by dividing the DNA polymorphisms according to their positions: genic regions, including coding sequence, introns, and untranslated regions, and non-genic regions, including intragenic spaces and 1-kb upstream regions (Table 6). The 1-kb upstream region exhibited the highest diversity for the common polymorphisms. For the uncommon polymorphisms, numerous SNPs, which were approximately 2.6-fold higher in EMT1 (136,821 SNPs) than in WT (52,741 SNPs) were found, whereas similar frequencies of InDels were observed in both the plants (24,991 vs. 22,599 for WT and EMT1, respectively). Similarly, the upstream region of SNPs showed the highest polymorphism frequency in both the plants. The start codon was changed in a total of 8 genes in EMT1, while it was changed in only one gene of WT.
The structural variations in EMT1 caused by γ-ray irradiation were evaluated by aligning the assembled EMT1 genome sequence with that of WT by using GASVpro-HQ with the default parameters (Table 7). For all 2,609 clusters consisting of 26,449 paired reads, deletions were detected in EMT1 across 12 chromosomes. The highest deletion frequency of 341 clusters with 4,511 paired reads was observed on chromosome 11. In contrast, inversions were observed in 120 clusters of EMT1 with 1,192 paired reads. The highest number of inversions (23 clusters with 188 paired reads) was found on chromosome 12.
Next, the lengths of InDels and their frequency in WT and EMT1 as compared to those in Nipponbare were calculated (Supplementary Fig. 2). For the uncommon polymorphisms of WT, InDels ranged from −58 to 34 bp in length. Similarly, in EMT1, InDels ranged from −57 to 35 bp in length. For the common polymorphisms in both the plants, InDels ranged from −44 to 33 bp in length. Mononucleotide InDels exhibited the highest frequency in all the three groups.
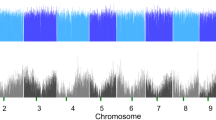
The frequency of SNPs and InDels and copy number variations (CNVs) at the chromosomal level were determined by visualizing their distributions on individual chromosomes as corresponding numbers within a 1-Mb interval of each chromosome (Fig. 2). Obvious variations in common polymorphisms were found in some regions across chromosomes, e.g., 712 SNPs/591 InDels within 10–17 Mb on chromosome 3. For the uncommon polymorphisms, differences in regions with variation and their frequency were also found compared to those in the reference genome. For example, uncommon polymorphisms were highly frequent in WT within 2–3 Mb on chromosome 1, with 1,619 SNPs/934 InDels whereas, in EMT1, they were found within 23–24 Mb on chromosome 3, with 5,666 SNPs/882 InDels. The distribution of CNVs in EMT1 and WT appeared to be highly consistent with the patterns of dense SNPs and InDels. The high density of CNVs on each chromosome was consistent with the dense regions of the uncommon polymorphisms in WT, whereas a low density was found in the dense regions of the uncommon polymorphisms in EMT1. For example, the highly dense region of the uncommon SNPs/InDels of EMT1 within 8–12 Mb on chromosome 6 exhibited an evidently lower number of CNVs, while the highly dense region of the common SNPs/InDels within 2–3 Mb on chromosome 1 clearly showed a high number of CNVs.

Distributions and frequencies of SNPs/InDels, CNVs, and unmapped genes. The x-axis represents the physical distance along each chromosome. The y-axis of SNPs and InDels show the numbers of polymorphisms. The frequency of SNPs and InDels was plotted as the corresponding numbers within a 1-Mb interval of each chromosome. The y-axis of CNV analysis indicates the log2 value
The genes with definite alterations in EMT1 compared to those in WT were determined by generating the WT consensus sequence by using SAMtool, and then the short reads of EMT1 were aligned on this sequence by using SOAPdenovo. After the unmapped reads of EMT1 were retrieved, they were subsequently aligned on the reference sequence. Interestingly, 40.73 % of the unmapped reads were matched to coding sequences (CDSs), corresponding to 1,275 genes, while the remaining reads were assigned into non-CDSs.
Transcriptome analysis of EMT1 genes with SNPs, InDels, and unmapped reads
Transcriptome analysis of EMT1 genes with sequence variations compared to those found in WT was performed by conducting microarray analyses by using leaf and panicle tissues from WT and EMT1. The WT and/or EMT1 genes, including SNPs, InDels, and unmapped reads, were retrieved, and their expression was determined on the basis of the microarray data (Fig. 3).

Gene numbers with SNPs and InDels and their transcriptome analysis. a Dendrograms of numbers of genes with SNPs and InDels, respectively. b Heatmaps of differentially expressed genes with SNPs and InDels and unmapped genes in EMT1. The red dots indicate the genes with the biological processes of flower development and growth. The red and green colors indicate up-regulation (log2 FC > 1) and down-regulation (log2 FC < −1), respectively, in EMT1 compared to of the expression in WT. (Color figure online)
In all, 706 genes harbored one or more SNPs compared to those in Nipponbare. A total of 302 genes showed common SNPs in both the plants, including 107 and 297 genes in WT and EMT1, respectively (Fig. 3a). In the case of InDels, 633 genes harbored at least one InDel in both the plants (389 genes) or in only EMT1 (233 genes) compared to those in the reference sequence (Fig. 3a).
Next, functional enrichment analysis of the retrieved EMT1 genes was performed on the basis of three Gene Ontology (GO) classifications, i.e., biological processes, molecular functions, and cellular components. The EMT1 genes were divided into three groups, i.e., common genes, uncommon genes, and unmapped genes. For the EMT1 genes with common SNPs, functional enrichment analysis indicated significant differences in certain biological functions such as flower development, signal transduction, and protein modification process (Supplementary Fig. 3a). In addition, the EMT1 genes with common SNPs were associated with functional enrichments in some molecular functions such as kinase activity and receptor activity and binding and in certain cellular components such as mitochondria and nucleus (Supplementary Fig. 3b and c). The EMT1 genes with uncommon SNPs were related with some biological functions, including flower development, whereas no such functional enrichments were observed for molecular functions or cellular components (Supplementary Fig. 4). In addition, the molecular functions of the common and uncommon InDels of EMT1 were represented by enzyme regulator activity and protein binding, respectively, whereas no such functional enrichments were observed for biological processes or cellular components (Supplementary Fig. 5a and b).
Whether the expression of the structurally changed genes in EMT1 was different compared with those of WT was also determined. The expression patterns in leaf and panicle tissues of WT and EMT1 were assessed using microarray analysis. A total of 181 genes, which consisted of 51 genes with common (20 genes) or uncommon SNPs (31 genes), 23 genes with common (20 genes) or uncommon InDels (3 genes), and 108 unmapped genes, showed differential expression patterns (twofold > or < −2-fold) in the leaves or panicles of EMT1 compared to those of WT. A total of 17 genes were associated with biological processes such as flower development (Fig. 3b).
A case study: molecular dissection of a mutated gene
The functions of the mutated genes were determined by using a gene (Os11g40890) encoding a putative LRR-RLK, which was isolated as a polymorphic fragment by using AFLP analysis. A full-length clone was obtained using GenomeWalker, and its sequence was confirmed using the resequencing data; the total length was 4,832 bp (full-length genomic sequence of 3,110 bp and upstream region of 1,722 bp). Alignment of the mutated gene (mOs11g40890) sequence and the EMT1 consensus sequence obtained from the resequencing data showed a high identity of 99.99 % (data not shown). In contrast, the consensus sequence of WT exhibited no difference with that of the reference genome. In addition, the sequences between AFLP fragments and the consensus sequences showed less than 1 % differences (data not shown), which might be caused by sequence errors or analysis of different generations.
The structure of the mutated gene was predicted using FGENESH (http://linux1.softberry.com/berry.phtml), and its predicted CDSs (Supplementary Fig. 6) and amino acid sequences (Fig. 4a) were aligned with the corresponding sequences of WT. Comparison of both alleles showed severe structural changes of the mutated gene, including a deletion block in the protein kinase domain, even though the C-terminal region was well conserved. The mutated gene encoded a 283-amino acid protein with a predicted molecular mass of 31.22 kDa and a calculated pI of 4.72, whereas the WT allele encoded a 793-amino acid protein with a molecular mass of 86.63 kDa and a calculated pI of 6.39. The expression patterns of each allele in WT and EMT1 were determined in the leaf and panicle tissues from each plant. Interestingly, the mutated gene was highly expressed in the leaves, but slightly induced in the panicles in EMT1, whereas the gene was not expressed in either tissue in WT (Fig. 4b). The subcellular localization of both the genes was identified, and their full-length genes were isolated from each plant. However, the gene from WT could not be expressed in any stressed leaves, including salt-, heat-, cold-, and drought-treated, as well as in healthy tissues, including leaves, roots, and panicles. These results might suggest that the WT gene is rarely expressed or not expressed at all (data not shown). Next, whether the protein of mOs11g40890 possesses protein kinase activity was determined. However, the protein did not exhibit kinase activity, while the Arabidopsis BRI1 protein showed kinase activity (Fig. 4c). A 35S::mOs11g40890-EGFP construct was generated that was expressed in tobacco leaves via agro-infiltration. The 35S::mOs11g40890-EGFP fluorescence signals were clearly observed in the cytosol. Similarly, 35::EGFP was also detected in the cytosol (Fig. 4d).

Molecular analysis of the mutated gene. a Amino acids alignment between wild type and mutated genes. b RT-PCR of both genes in each leaf and panicle of both plants. c In vitro kinase assay of the mutated gene. The Arabidopsis BRI1 protein was used as a positive control. d Subcelluar localization of the mutated gene. The 35S::EGFP and 35S::mOs11g40890-EGFP constructs were transiently expressed via agro-infiltration in tobacco leaves for 3 days
Discussion
Non-transgenic TILLING method for reverse genetics is generally believed to be useful for the investigation of novel genetic variations and has also been used to improve crop plants (Slade and Knauf 2005). In addition, mutagenesis has been utilized extensively to create genetic diversity in conventional breeding and further used for powerful reverse genetic strategies such as TILLING (Perry et al. 2009). Our TILLING population might be a useful genetic material pool for application in mutation breeding as well as reverse genetic approaches (Subbaiyan et al. 2012). This study provides evidence that the EMT lines show not only an early heading date but also excellent agronomic traits regarding yield potential such as grains per panicle and seed setting rate.
Due to recent rapid advances in next-generation sequencing, many rice cultivars have been sequenced using whole-genome resequencing, e.g., the elite Japanese rice cultivar Koshihikari (Yamamoto et al. 2010), the landrace japonica cultivar Omachi (Arai-Kichise et al. 2011), 6 elite indica rice inbreds (Subbaiyan et al. 2012), and three elite restorer lines (Li et al. 2012). The pool of available DNA polymorphisms could be utilized for genetic analysis and to improve rice varieties (Subbaiyan et al. 2012). However, a genome-wide approach for discovering DNA polymorphisms between a WT plant and its mutant has not yet been reported in rice. This approach could accelerate the molecular dissection of the mutated phenotypes and molecular breeding via gene pyramiding.
The SNP densities between Nipponbare and WT and between Nipponbare and EMT1 were 140.8 SNPs and 365.5 SNPs per Mb, respectively. The density between Nipponbare and WT was somewhat similar to the average of 175 SNPs per Mb between Nipponbare and its closely related cultivar, Koshihikari (Yamamoto et al. 2010), whereas the density between Nipponbare and EMT1 was similar to the value of 346.6 SNPs per Mb between Nipponbare and a landrace cultivar (Arai-Kichise et al. 2011). These results allowed the speculation about the degree of genetic alteration created via ionizing radiations such as γ-rays.
The finding that the transition to transversion ratio was 2.23 and 2.30 for the uncommon SNPs of WT and EMT1, respectively, and 2.53 for the common SNPs supports the hypothesis regarding transition bias (Morton 1995) and is consistent with the results from the six elite indica rice inbreds (Subbaiyan et al. 2012). Interestingly, the frequency of transitions and transversions in the uncommon SNPs of EMT1 were considerably higher (1.75- to 4.85-fold) than those in WT, supporting the hypothesis that γ-ray irradiation generated extreme DNA alterations in EMT1.
The finding regarding the high frequency of non-synonymous substitutions in both plants compared to that of synonymous substitutions (Table 6) might be unexpected, because the rate of synonymous substitutions (silent) is generally considerably higher than that of non-synonymous substitutions (amino acid alterations; Nei and Gojobori 1986). However, a similar finding was observed when the six elite indica rice inbreds were compared to the genome of Nipponbare (Subbaiyan et al. 2012). Further studies are warranted to explain the unexpected results.
We determined whether the genes related to the photoperiodic pathway controlling flowering time were significantly altered in the EMT1 genome (Mouradov et al. 2002). A total of 17 rice genes related to the photoperiodic pathway were retrieved from the KEGG database. Interestingly, only two genes encoding a signal transduction response regulator (Os02g40510 and Os07g49460) showed a nonsynonymous substitution in EMT1, whereas 1 gene (Os02g40510) exhibited a nonsynonymous substitution in WT compared to that in the Nipponbare genome (Supplementary Table 3). This finding might suggest that the early heading date of the EMT1 line was not correlated to the circadian rhythm pathway genes, but to genes of other floral pathways. An alternative hypothesis is that the phenotype was caused by the changes in the transcriptional regulation of the genes through cis-regulatory alterations. The finding that differences in the expression patterns of photoperiodic pathway genes (Supplementary Table 3) were observed in EMT1 compared to those of WT might support this hypothesis.
Structural alterations of genes are generally believed to modify their function and expression. We found that at least 202 structurally altered genes showed changes in expression in EMT1, and functional enrichment analysis of these genes showed that their molecular functions were related to flower development. Although further studies are required to identify the specific gene(s) responsible for the early heading date, the genes identified in our study could be interesting candidates for investigation in further studies. A representative example is the mutated gene (mOs11g40890) encoding a putative LRR-RLK, which was highly inducible in the leaves of EMT1 (but not of WT) and localized to the cytosol (Fig. 4). The large LRR-RLK family includes at least 292 members in rice genomes, and their functional diversity has been proposed during evolution (Hwang et al. 2011); plant LRR-RLKs are believed to regulate plant development, including flowering organ abscission (Diévart and Clark 2004). Although the exact function of the mutated gene, especially with respect to flowering, was not determined, this gene might be an interesting candidate for further studies.
We selected EMT lines from a TILLING population and conducted whole-genome resequencing to identify genome-wide DNA polymorphisms and microarray analysis to verify the expression patterns of the structurally altered genes. Our results might provide a critical insight on the regulatory pathways associated with rice flowering and help improve our knowledge regarding mutation breeding.
Abbreviations
- TILLING:
-
Targeting-induced local lesions in genomes
- EMT:
-
Early-maturing TILLING
- WT:
-
Wild type
- LRR-RLK:
-
Leucine-rich repeat receptor-like kinase
- Hd1 :
-
Heading date 1
- Ehd1 :
-
Early heading date 1
- Ghd7 :
-
Grain number, plant height, and heading date 7
- BAM:
-
Binary format
- SAM:
-
Sequence alignment map
- SNP:
-
Single nucleotide polymorphism
- InDels:
-
Insertions and deletions
- SVs:
-
Structural variations
- GO:
-
Gene ontology
- CTAB:
-
Cetyltrimethylammonium bromide
- AFLP:
-
Amplified fragment length polymorphism
References
Arai-Kichise Y, Shiwa Y, Nagasaki H, Ebana K, Yoshikawa H, Yano M, Wakasa K (2011) Discovery of genome-wide DNA polymorphisms in a landrace cultivar of japonica rice by whole-genome sequencing. Plant Cell Physiol 52:274–282
Cho HY, Park SJ, Kim DS, Jang CS (2010) A TILLING rice population induced by gamma-ray irradiation and its genetic diversity. Korean J Breed Sci 42:365–373
Diévart A, Clark SE (2004) LRR-containing receptors regulating plant development and defense. Development 131:251–261
Doi K, Izawa T, Fuse T, Yamanouchi U, Kubo T, Shimatani Z, Yano M, Yoshimura A (2004) Ehd1, a B-type response regulator in rice, confers short-day promotion of flowering and controls FT-like gene expression independently of Hd1. Genes Dev 18:926–936
Higgins JA, Bailey PC, Laurie DA (2010) Comparative genomics of flowering time pathways using Brachypodium distachyon as a model for the temperate grasses. PLoS ONE 5:e10065
Hwang SG, Kim DS, Jang CS (2011) Comparative analysis of evolutionary dynamics of genes encoding leucine-rich repeat receptor-like kinase between rice and Arabidopsis. Genetica 139:1023–1032
Jones N, Ougham H, Thomas H, Pasakinskiene I (2009) Markers and mapping revisited: finding your gene. New Phytol 183:935–966
Kim DS, Lee IS, Jang CS, Lee SJ, Song HS, Lee YI, Seo YW (2004a) AEC resistant rice mutants induced by gamma-ray irradiation may include both elevated lysine production and increased activity of stress related enzymes. Plant Sci 167:305–316
Kim DS, Lee CS, Jang CS, Hyun DY, Seo YW, Lee YI (2004b) Selection of 5-methyltryptophan resistant rice mutants from irradiated calli derived from embryos. Euphytica 135:9–19
Li H, Durbin R (2010) Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26(5):589–595
Li J, Wen J, Lease KA, Doke JT, Tax FE, Walker JC (2002) BAK1, an Arabidopsis LRR receptor-like protein kinase, interacts with BRI1 and modulates brassinosteroid signaling. Cell 26:213–222
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup (2009) The sequence alignment/map format and SAMtools. Bioinformatics 15(25):2078–2079
Li S, Wang S, Deng Q, Zheng A, Zhu J, Liu H, Wang L, Gao F, Zou T, Huang B, Cao X, Xu L, Yu C, Ai P, Li P (2012) Identification of genome-wide variations among three elite restorer lines for hybrid-rice. PLoS ONE 7(2):e30952
Lim SD, Yim WC, Moon JC, Kim DS, Lee BM, Jang CS (2010) A gene family encoding RING finger proteins in rice: their expansion, expression diversity, and co-expressed genes. Plant Mol Biol 72(4–5):369–380
Morton BR (1995) Neighboring base composition and transversion/transition bias in a comparison of rice and maize chloroplast noncoding regions. Proc Natl Acad Sci USA 106:12273–12278
Mouradov A, Cremer F, Coupland G (2002) Control of flowering time: interacting pathways as a basis for diversity. Plant Cell 14:S111–S130
Nei M, Gojobori T (1986) Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol 3:418–426
Patel RK, Jain M (2012) NGS QC toolkit: a toolkit for quality control of next generation sequencing data. PLoS ONE 7(2):e30619
Perry J, Brachmann A, Welham T, Binder A, Charpentier M, Groth M, Haage K, Markmann K, Wang TL, Parniske M (2009) TILLING in Lotus japonicus identified large allelic series for symbiosis genes and revealed a bias in functionally defective ethyl methanesulfonate alleles toward glycine replacements. Plant Physiol 151(3):1281–1291
Saghai-Maroof MA, Soliman KM, Jorgensen RA, Allard RW (1984) Ribosomal DNA spacer-length polymorphisms in barley: Mendelian inheritance, chromosomal location, and population dynamics. Proc Natl Acad Sci USA 81:8014–8018
Shin MS, Ko JK, Kim BK, Lee KS, Lee JK, Ha KY, Yang BK, Shin HT, Lee SY, Kim CH, Cho SY (1997) A new high yielding and good quality rice cultivar adaptable to direct seeding “Donganbyeo”. Korean J Breed Sci 29:495
Sindi S, Onal S, Peng L, Wu H, Raphael BJ (2012) An integrative model for identification of structural variation in sequencing data. Genome Biol 13:R22
Slade AJ, Knauf VC (2005) TILLING moves beyond functional genomics into crop improvement. Transgenic Res 14(2):109–115
Subbaiyan GK, Waters DL, Katiyar SK, Sadananda AR, Vaddadi S, Henry RJ (2012) Genome-wide DNA polymorphisms in elite indica rice inbreds discovered by whole-genome sequencing. Plant Biotechnol J 10(6):623–634
Till BJ, Reynolds SH, Greene EA, Codomo CA, Enns LC, Johnson JE, Burtner C, Odden AR, Young K, Taylor NE, Henikoff JG, Comai L, Henikoff S (2003) Large-scale discovery of induced point mutations with high-throughput TILLING. Genome Res 13:524–530
Vos P, Hogers R, Bleeker M, Reijans M, van de Lee T, Frijters A, Pot J, Peleman J, Kuiper M (1995) AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res 23:4407–4414
Xue W, Xing Y, Weng X, Zhao Y, Tang W, Wang L, Zhou H, Yu S, Xu C, Li X, Zhang Q (2008) Natural variation in Ghd7 is an important regulator of heading date and yield potential in rice. Nat Genet 40:761–767
Yamamoto T, Nagasaki H, Yonemaru J, Ebana K, Nakajima M, Shibaya T, Yano M (2010) Fine definition of the pedigree haplotypes of closely related rice cultivars by means of genome-wide discovery of single-nucleotide polymorphisms. BMC Genom 11:267
Yano M, Katayose Y, Ashikari M, Yamanouchi U, Monna L, Fuse T, Baba T, Yamamoto K, Umehara Y, Nagamura Y, Sasaki T (2000) Hd1, a major photoperiod sensitivity quantitative trait locus in rice, is closely related to the Arabidopsis flowering time gene CONSTANS. Plant Cell 12:2473–2483
Acknowledgments
This work was supported by grants from iPET (Korea Institute of Planning and Evaluation for Technology in Food, Agriculture, Forestry and Fisheries), Ministry for Food, Agriculture, Forestry and Fisheries and the Next-Generation BioGreen 21 Program (Plant Molecular Breeding Center No. PJ009084), Rural Development Administration, Republic of Korea and 2013 Research Grant from Kangwon National University (No. 120131456).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Hwang, SG., Hwang, J.G., Kim, D.S. et al. Genome-wide DNA polymorphism and transcriptome analysis of an early-maturing rice mutant. Genetica 142, 73–85 (2014). https://doi.org/10.1007/s10709-013-9755-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10709-013-9755-0