
Abstract
Long terminal repeat (LTR) retrotransposons make up substantial parts of most higher plant genomes where they accumulate due to their replicative mode of transposition. Although the transposition is facilitated by proteins encoded within the gag-pol region which is common to all autonomous elements, some LTR retrotransposons were found to potentially carry an additional protein coding capacity represented by extra open reading frames located upstream or downstream of gag-pol. In this study, we performed a comprehensive in silico survey and comparative analysis of these extra open reading frames (ORFs) in the group of Ty3/gypsy LTR retrotransposons as the first step towards our understanding of their origin and function. We found that extra ORFs occur in all three major lineages of plant Ty3/gypsy elements, being the most frequent in the Tat lineage where most (77 %) of identified elements contained extra ORFs. This lineage was also characterized by the highest diversity of extra ORF arrangement (position and orientation) within the elements. On the other hand, all of these ORFs could be classified into only two broad groups based on their mutual similarities or the presence of short conserved motifs in their inferred protein sequences. In the Athila lineage, the extra ORFs were confined to the element 3′ regions but they displayed much higher sequence diversity compared to those found in Tat. In the lineage of Chromoviruses the extra ORFs were relatively rare, occurring only in 5′ regions of a group of elements present in a single plant family (Poaceae). In all three lineages, most extra ORFs lacked sequence similarities to characterized gene sequences or functional protein domains, except for two Athila-like elements with similarities to LOGL4 gene and part of the Chromoviruses extra ORFs that displayed partial similarity to histone H3 gene. Thus, in these cases the extra ORFs most likely originated by transduction or recombination of cellular gene sequences. In addition, the protein domain which is otherwise associated with DNA transposons have been detected in part of the Tat-like extra ORFs, pointing to their origin from an insertion event of a mobile element.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In higher plants, long terminal repeat (LTR) retrotransposons represent a major fraction of repetitive DNA. Even small plant genomes such as those of Arabidopsis or rice contain 5.6–17 % of LTR retrotransposons and this proportion increases along with increasing genome size (Pereira 2004; McCarthy et al. 2002; Zuccolo et al. 2007). Amplification of a single family of elements can lead up to a 60 % increase in nuclear DNA content (Neumann et al. 2006) and it has been well documented that differential accumulation of LTR retrotransposons is one of the key forces causing an extraordinary variation in genome size observed in higher plants (Hawkins et al. 2008). The genomic accumulation of LTR retrotransposons is due to their replicative mode of transposition involving transcription of the parental element, reverse transcription of the resulting RNA into cDNA and subsequent integration of the new element copy into the genome. However, it is not known why LTR retrotransposons are so successful in colonizing plant genomes compared to other types of retroelements like LINEs or SINEs that use the same mode of transposition.
The replication of LTR retrotransposons is facilitated by a set of proteins encoded by the gag-pol sequence located in the internal region between the two direct terminal repeats. The gag gene codes for proteins needed for the assembly of virus-like particles and RNA packaging. The pol gene encodes enzymes protease (Pro), reverse transcriptase/RNaseH (RT/RH) and integrase (INT). RT/RH and INT convert retrotransposon RNA into DNA and integrate it into the genome, respectively. The order of RT/RH–INT domains within the pol gene is typical for Ty3/gypsy elements, while the other major group of LTR retrotransposons, Ty1/copia, has the INT domain located upstream of the RT/RH. Translation of the gag-pol region occurs from a single open reading frame (ORF) and individual functional proteins are released from a precursor polyprotein by the action of protease (Kumar and Bennetzen 1999; Havecker et al. 2004). Alternatively, in some groups of elements the gag-pol region contains several overlapping or adjacent ORFs and its translation is facilitated by translational recoding mechanisms including ribosomal frameshifting and stop codon bypass (Gao et al. 2003; Forbes et al. 2007) or by RNA splicing (Steinbauerová et al. 2008).
While the gag-pol-encoded proteins are considered sufficient to accomplish the LTR retrotransposon replication and transposition, a number of elements have been found to carry an additional protein coding capacity. For example, fragments of ATPase, 1,4-β-xylan endohydrolase and 1,3-β-glucanase sequences were identified within the maize element Bs1 (Jin and Bennetzen 1994; Elrouby and Bureau 2001), demonstrating the ability of plant retrotransposons to transduce cellular genes. In these cases, the gene sequences were found to generate fusion open reading frames with the truncated gag sequences. However, in several groups of elements there are additional reading frames that are separate from the gag-pol region (hereafter termed “extra ORFs” or “eORFs”). They occur in elements from two of the seven evolutionary lineages defined for Ty1/copia (Wicker and Keller 2007) and appear to be even more frequent and diverse in Ty3/gypsy elements, differing in their location (5′ or 3′ from the gag-pol) as well as in their orientation within the elements. The best documented are the eORFs located at 3′ regions of elements from the Athila lineage that were suggested to encode Env-like proteins analogous to retroviral env genes (Peterson-Burch et al. 2000; Wright and Voytas 2002). Whereas these eORFs are in the same orientation as the gag-pol genes, several elements were identified including Retand from Silene latifolia, RIRE2 from rice or Grande1 from maize that contained ORFs located 3′ of pol and in the opposite (antisense) orientation (Kejnovsky et al. 2006; Ohtsubo et al. 1999; Martínez-Izquierdo et al. 1997). These elements belong to the Tat lineage of Ty3/gypsy retrotransposons, which also includes a group of Ogre retrotransposons with eORFs located at 5′ sequence regions (Neumann et al. 2003; Macas and Neumann 2007). The same position was reported for eORFs in the rice RIRE3 and RIRE8 elements representing the lineage of plant Chromoviruses (Kumekawa et al. 1999).
Most of the eORFs identified so far showed no detectable similarity to known genes. Moreover, it should be noted that their nucleotide sequences are relatively heterogeneous, and it is mainly their location that appears to be conserved in some Ty3/gypsy families. On the other hand, the repeated presence of eORFs in groups of related elements raise questions about their origin and eventual evolutionary importance. These questions have been difficult to address, however, due to only scattered information about the occurrence and sequence composition of the eORFs. Therefore, in this work we performed a systematic in silico survey of eORFs in Ty3/gypsy elements identified in available seed plant (Spermatophyta) genomic sequence data. The elements were detected and classified based on their structure and sequence similarities, and a number of novel elements containing eORFs were identified in all three Ty3/gypsy evolutionary lineages (Tat, Athila and Chromovirus; Lloréns et al. 2008). A comparative analysis of nucleotide and putative protein sequences of all eORFs was performed and analyzed in the context of element structure and evolution.
Methods
Input data
Seed plant (Spermatophyta) genomic DNA sequences used for analysis were obtained from GenBank and from plant genome sequencing projects listed below. In summary, the available sequence data accounted for 1.6 Mbp for gymnosperms, 0.12 Mbp for magnoliids, 8.5 Gbp for eudicotyledons and 4.1 Gbp for monocotyledons (99.9 % of which were from Poaceae) taxa. The GenBank sequences (7,230 entries) were downloaded via NCBI Entrez server (http://www.ncbi.nlm.nih.gov/entrez/) and were limited to sequences at least 5 kb long, excluding data from species that were downloaded as whole genome sequencing projects. These included Arabidopsis thaliana (TAIR release 9, http://www.arabidopsis.org/), Eucalyptus grandis (v1.0 8X, http://eucalyptusdb.bi.up.ac.za/), Medicago truncatula (Medicago_3.0, http://medicago.org/genome), Nicotiana tabacum (http://www.pngg.org/tgi/), Solanum lycopersicum (v2.30, http://mips.helmholtz-muenchen.de/plant/tomato/), and the following species that were downloaded via Phytozome project web (http://www.phytozome.net): Aquilegia coeruela (8x unmapped genome assembly), Arabidopsis lyrata (JGI v1.0; Hu et al. 2011), Brachypodium distachyon (GI v1.0 8x, International Brachypodium Initiative 2010), Carica papaya (Ming et al. 2008), Cucumis sativus (Csativus_122), Glycine max (Glyma1; Schmutz et al. 2010), Manihot esculenta (Cassava4), Mimulus guttatus (v1.0), Oryza sativa (MSU 6.0; Ouyang et al. 2007), Populus trichocarpa (JGI v2; Tuskan et al. 2006), Prunus persica (v1.0), Ricinus communis (TIGR/JCVI v0.1), Setaria italica (v1 8.3x), Sorghum bicolor (v1.0; Paterson et al. 2009), Vitis vinifera (March 2010 release 12x; Jaillon et al. 2007), Zea mays (4a.53; Schnable et al. 2009).
Identification of Ty3/gypsy LTR retrotransposons
Computer analyses were performed using custom BioPerl (http://www.bioperl.org/) and R (http://www.r-project.org/) scripts or the external programs specified below run on a Debian Linux server (16 CPUs, 72 GB RAM). Identification of all types of intact LTR retrotransposons was done using LTR_FINDER (Xu and Wang 2007) with the following parameters differing from the program defaults: maximum length of LTRs set to 7 kb, LTRs had to include terminal “TG” and “CA” dinucleotides, PBS detection threshold (minimal tRNA match) was 12 bp and target site duplications were required to surround the elements. The identified LTR retrotransposon sequences were subjected to FASTY (Pearson et al. 1997) similarity search against our comprehensive database of Gag-Pol protein domains derived from elements representing all major lineages of plant retrotransposons (Wicker and Keller 2007; Lloréns et al. 2008). The sequences that gave hits of at least 50 % similarity over 80 % of domain length to all three, Gag, RT and INT domains and produced the best hits to entries from Ty3/gypsy retrotransposons were selected as Ty3/gypsy elements. These elements were further processed using the cd-hit program (Li et al. 2001, 2002) in order to detect and eliminate identical elements that originated from duplicated sequences present in the analyzed input DNA data.
Detection and analysis of extra ORFs
Open reading frames of at least 300 bp in length were identified in retrotransposon internal sequences (the regions located between the LTRs) using the getorf program (EMBOSS, http://emboss.sourceforge.net/) and examined for their location relatively to the ORFs containing Gag and INT domains within the same element. All ORFs (regardless of their frame and orientation) located upstream of the Gag-containing ORF were collected as 5′ eORFs, whereas those located downstream of the ORF including INT domain were designated 3′ eORFs. Special care was taken to exclude false 5′ eORFs that originated by nonsense or frameshift mutations within a relatively variable 5′ end of the Gag-coding region. These cases were revealed by comparison to previously characterized full-length gag genes. In addition, ORFs present at sequence regions comprised of tandem repeats were identified using Tandem Repeats Finder (Benson 1999) and excluded because their occurrence was due to the lack of stop codons in these low complexity sequence regions.
In order to efficiently handle the large numbers and sequence diversity of identified eORFs, their sequences were subjected to the similarity-based clustering analysis as described by Novák et al. (2010). This procedure employs graph-based representation of sequence similarities, that results in the clustering of overlapping sequences and allows one to identify representatives for each group according to their high numbers of overlaps to other sequences within the same group. These statistics were combined with a manual examination of the cluster graphs using SeqGrapheR program (Novák et al. 2010), resulting in the selection of a limited set of typical eORFs (Online Resource 1) as well as the corresponding elements of which they were found for further analysis (complete elements are provided in Online Resource 2 and their internal regions in Online Resource 3). Newly identified elements were labeled by abbreviated species name and number (for example, Sb1 is the element #1 from Sorghum bicolor) while previously described elements are shown under their respective names. Phylogenetic relationships of the selected elements including their assignment to basic lineages of Ty3/gypsy retrotransposons (Lloréns et al. 2008) were assessed based on sequence similarities of their RT domains as described by Neumann et al. (2011). Phylogenetic trees presented on Figs. 1, 3, 4 and 5 were solely based on this analysis of RT domain similarities.

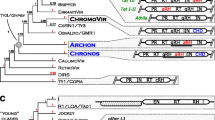
Distribution of eORFs in Ty3/gypsy elements. A phylogenetic tree inferred from a sequence comparison of reverse transcriptase domains shows three major lineages of Ty3/gypsy retrotransposons with the locations of eORFs upstream (5′) or downstream (3′) of the gag-pol region distinguished by color shading. The names show positions of previously described retrotransposons while all other branches represent the elements identified in this study
Similarities of protein sequences obtained by conceptual translations of eORFs were detected using FASTA (Pearson and Lipman 1988). In order to reveal subtle similarities or short conserved motifs in diverged sequences, eORFs were also examined using MEME and MAST programs (The MEME suite, http://meme.sdsc.edu/meme/; MEME parameters: motif width 5–60 amino acids, motif p value < 1e−05; MAST parameters: sequence E value < 0.1, motif p value < 0.0001). Similarities to previously defined conserved protein domains were detected by searching Pfam 26.0 (http://pfam.janelia.org/; Finn et al. 2010) and the Conserved Domain Database (CDD, http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi; Marchler-Bauer et al. 2011). Similarities to proteins in NCBI non-redundant protein sequences (nr) database and Swissprot protein sequences (Swissprot) database were detected using BLASTP (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Transmembrane domains were identified using TMpred with values >500 considered as significant (http://www.ch.embnet.org/software/TMPRED_form.html; Hofmann and Stoffel 1993).
Results
Identification and phylogenetic classification of plant Ty3/gypsy elements carrying extra ORFs
A total of 12.6 Gbp of sequence data representing seed plant (Spermatophyta) sequences available from GenBank and genome assemblies of extensively sequenced species was screened for LTR retrotransposons using LTR_FINDER program (Xu and Wang 2007). Over 88,000 potential elements were retrieved based on their structural and sequence features including the presence of intact LTRs, primer binding sites and target site duplications. Ty3/gypsy elements were then selected based on best similarities of their gag-pol regions to the database of conserved coding domains including representative sets of both, Ty1/copia and Ty3/gypsy sequences. Due to this selection procedure, only the elements containing complete coding regions were included, removing non-autonomous elements that lacked their gag-pol sequences. A set of 18,172 unique Ty3/gypsy elements was finally assembled and used for further analysis consisting of the identification of open reading frames of at least 300 bp located upstream (5′ eORFs) or downstream (3′ eORFs) of the gag-pol. Such eORFs were identified in all three lineages of Ty3/gypsy retrotransposons, being the most frequent in Tat where 77 % of elements carried eORFs (Table 1). The presence of eORFs was further investigated with respect to the phylogenetic relationships of the elements that were estimated by analyzing the similarities of their reverse transcriptase (RT) domains (Fig. 1). This analysis revealed that locations of eORFs are mostly conserved within groups (clades or families) of elements. For example, in Chromoviruses there were only 5′ eORFs detected which were confined to a subset of elements represented by the Tekay clade but missing in the Reina and CRM clades defined by Gorinsek et al. (2004). On the contrary, the location of eORFs in the Athila lineage was exclusively on the 3′ end. Both types of arrangements were identified in the Tat lineage where most retrotransposons included 3′ located eORFs whereas Ogre-like elements were characterized by eORF located upstream of the gag-pol or in both, 5′ and 3′ regions (Figs. 1, 2). As the eORFs shared varying degrees of sequence similarities, they were clustered to groups of similar sequences in order to identify typical elements for a more detailed analysis described below. The list of these representative elements is provided as Online Resource 4 along with their full-length nucleotide sequences (Online Resource 2) and protein sequences of corresponding eORFs (Online Resource 1).

Schematic representation of various types of eORF arrangements within Ty3/gypsy elements. Open reading frames are represented by rectangles and their positions above or below the lines correspond to their forward or reverse orientation, respectively. Sequence similarities between eORFs are highlighted by colors and hatching. Details about depicted elements can be found in the Online Resource 4, except for Ogre-SL which represents a consensus sequence reconstructed from Silene latifolia shotgun sequencing data (Macas et al. 2011)
Tat lineage
Of the three basic lineages of Ty3/gypsy retrotransposons, the Tat lineage was found to have the highest proportion of elements carrying eORFs (Table 1) as well as the highest variation in eORF arrangement within the elements (Fig. 2). There were four groups of elements categorized according to similarities in protein sequences obtained by conceptual translations of their eORFs, the occurrence of short conserved motifs in these sequences and phylogenetic relationships estimated from similarities of the element RT-coding domains (Fig. 3; Online Resource 5).

Extra ORFs identified in the Tat lineage of Ty3/gypsy elements. The elements are arranged based on their positions within a phylogenetic tree inferred from sequence similarities of their RT domains (left panel; bootstrap values are shown for the major nodes only). Newly identified elements are labeled by abbreviated species names and element numbers (for their details see Online Resource 4). The central panel is a dot-plot representation of mutual similarities of putative eORF protein sequences determined using FASTA (Pearson et al. 1997) and displayed as shades of gray according to the scale above the plot (darker color corresponds to higher similarity). The order of eORF sequences is the same along the horizontal and vertical axes of the plot. In the right panel, “+” marks the presence of various eORF features as specified above each column. The presence of eORFs in 5′ element regions and in forward orientation is distinguished from the 3′ and reverse orientation in the rest of the elements (blank space; the “−” mark in the case of Ogre-VV denotes the lack of any eORF). Furthermore, the presence of a plant mobile domain (PMD) and various short sequence motifs is also indicated (for motif sequences see Online Resource 5). Origin of the elements is provided in the “Taxonomy” panel, distinguishing eudicots (eud) and monocots (mon) and specifying plant families as follows: B Brassicaceae; C Caryophyllaceae; F Fabaceae; Ma Malvaceae; My Myrtaceae; Ph Phrymaceae; P Poaceae; Ra Ranunculaceae; Ro Rosaceae; Sa Salicaceae; So Solanaceae; V Vitaceae
Group A corresponded to the phylogenetically and structurally distinct clade of Ogre elements that is characterized by the conserved occurrence of forward-oriented 5′ eORFs in their sequences. The Ogres were previously identified in a variety of dicot species and were found to possess several specific features in addition to the eORFs, including the presence of an intron within their gag-pol region, primer binding sites with similarity to tRNAArg and extremely long LTRs (Macas and Neumann 2007). In the present study, we have identified Ogre-like elements in additional plant families (Malvaceae, Myrtaceae) and found detectable similarities between all Ogre 5′ eORF proteins, including those from relatively distant taxa (Fig. 3). No similarity to known gene sequences available from the NCBI and Swissprot protein databases was found that could clearly indicate the origin of Ogre eORFs. However, partial but significant (e value 1e−5–1e−23) similarities of most eORFs were detected to the plant mobile domain (PMD) by searching conserved domain databases Pfam and CDD (Finn et al. 2010; Marchler-Bauer et al. 2011). The PMDs are characterized by several conserved charged/polar residues and were reported to be associated with MULE transposases as well as being present as stand-alone domains in some plant genomes (Babu et al. 2006).
Although the characteristics described above were valid for a majority of group A elements, there were two notable exceptions. The first one was the previously described Glycine max element GmOgre (Laten et al. 2009) that in addition to a 5′ eORF included also a 3′ eORF (Fig. 2) with no detectable similarity to any other eORF from the Tat lineage (Fig. 3). However, similarities to 3′ eORFs of the elements from the Cyclops/Calypso group of the Athila lineage were detected (the highest similarity was to the element Gt1 with 38 % identities/46 % similarities of their predicted protein sequences). The second exception included the element Ogre-SL from Silene latifolia that lacked a 5′ ORF but contained a 3′ eORF in reverse orientation (Fig. 2) sharing protein similarity (including the presence of PMD) with the Ogre 5′ eORFs (Fig. 3).
Group B consisted of elements carrying 3′ eORFs in reverse orientation (Fig. 2) identified in diverse taxa including monocot and dicot species. Interestingly, these eORFs shared similarities to PMD and protein motifs 1 and 2 with 5′ eORFs from group A (Fig. 3) that points to a common origin. The groups C and D elements also contained reverse-oriented 3′ eORFs, but they had no similarity to those in groups A and B. On the other hand, elements of groups C and D shared three short protein motifs and similarity to the CDD database entry pfam04195 (gypsy-related protein domain). Sequence diversity of eORFs differed between these groups, being higher in C than in D. However, this difference could be at least in part due to a generally higher sequence similarity of group D elements that mostly occurred in closely related taxa (Poaceae family, with the exception of Silene latifolia element Retand, Fig. 3). It should be noted that in both groups there was considerable variability in eORF length, probably due to mutation-caused fragmentation of originally longer reading frames.
Athila lineage
In this lineage, eORFs were located in 3′ regions and oriented in a forward direction (Fig. 2), except for the reverse-oriented eORF of the Pp1 element. The eORFs occurred in elements from a wide range of species including gymnosperm, dicot and monocot plants (Fig. 4). There was considerable diversity revealed in eORF putative protein sequences, with no sequences or short motifs found to be conserved across all elements. This feature was best evident for a group of closely related Sorghum bicolor elements Sb9, Sb10 and Sb11 with highly similar RT domain sequences but no similarities between their eORFs. On the other hand, sequence similarities and shared motifs were observed within some groups including elements from different species. In the case of the Athila4 clade, there was similarity detected to the conserved domain pfam03078 that has been previously proposed as a typical motif of Athila-like eORFs. The majority of eORFs lacked any similarity to known gene sequences, however, significant similarities (e values of 6e−66 and 6e−49, respectively) were detected for Pt1 and Cs1 elements to A. thaliana LOGL4 protein (cytokinin riboside 5′-monophosphate phosphohydrolase; Kuroha et al. 2009). As the eORFs in Athila-like elements are considered analogous to retroviral env genes, we searched their inferred protein sequences for the presence of transmembrane domains that are characteristic for Env-like proteins (Wright and Voytas 2002). Although the transmembrane domains were predicted in most eORFs, the predictions fell below the threshold score for several groups of elements (Fig. 4).

Extra ORFs identified in the Athila lineage. The figure layout is as described for Fig. 3. In this lineage, eORFs were located in 3′ regions and arranged in forward orientation, except for Pp1 (eORF in reverse orientation). In the right panel, presence of short protein motifs (specified in Online Resource 5) in eORFs is indicated, as well as the similarity to LOGL4 gene and the presence of putative transmembrane domains (column “TM domain”). Taxonomy: gym, gymnosperms; eud eudicots; mon monocots; A Amaranthaceae; B Brassicaceae; E Euphorbiaceae; F Fabaceae; Ma Malvaceae; Ph Phrymaceae; Pi Pinaceae; P Poaceae; Ra Ranunculaceae; Ro Rosaceae; Ru Rutaceae; Sa Salicaceae; So Solanaceae; V Vitaceae
Chromoviruses
In the lineage of plant Chromoviruses, the identified eORFs were confined to 5′ regions of the elements and were in the same (forward) orientation as the gag-pol ORF (Fig. 2). Their occurrence was limited to a subset of elements within the Tekay clade identified in grass (Poaceae) genomes including previously described retrotransposons Retrosat2, RIRE3, RIRE8 and FRetro3 (Kumekawa et al. 1999; Gao et al. 2009). On the other hand, no eORFs were detected in the clades of centromeric (CRM) and Reina-like Chromoviruses (Fig. 5). The identified eORFs showed varying degrees of mutual similarities of their putative protein sequences, including a set of five conserved sequence motifs with scattered occurrence across all eORFs, suggesting a common origin. Significant partial sequence similarity was found for a subset of eORFs to 24 residues representing the N-terminal end of the histone H3 proteins (Fig. 5). In a part of the eORFs proteins, this similarity also corresponded to their N-terminal regions, whereas it was located internally in the rest of eORFs (Fig. 6). It should be noted that although the similarity to H3 varied between individual eORFs, there were some conserved positions that corresponded to the residues which are frequently targeted by epigenetic histone H3 modifications, including lysine K9 (Fig. 6).

Extra ORFs identified in the Chromovirus lineage. The figure layout is as described for Fig. 3. In this lineage, all eORFs were located in 5′ regions and arranged in forward orientation. In the right panel, presence of short protein motifs (specified in Online Resource 5) in eORFs is indicated. All eORFs with partial similarity to histone H3 are indicated in the column “H3”, while eORFs where this similarity corresponded to their N-terminal regions are marked in the column “N-terminal H3”. Alignment of these sequences is provided in Fig. 6. Taxonomy: eud eudicots; mon monocots; B Brassicaceae; F Fabaceae; P Poaceae; So Solanaceae

Alignment of the N-terminal region of A. thaliana histone H3 protein with protein sequences inferred from 5′ eORFs of selected Chromovirus elements. The residues targeted by epigenetic modifications are labeled by M (methylation), A (acetylation) and P (phosphorylation) according to Loidl (2004) and UniProt database (http://www.uniprot.org/uniprot/P59226)
Discussion
A systematic survey of plant Ty3/gypsy elements performed in this study revealed that they frequently contain extra open reading frames in addition to the common gag-pol region and that the positions of these eORFs are conserved in groups of related elements. Thanks to the availability of sequence data from a wide range of plant taxa it was also possible to investigate the diversity of eORFs present in elements belonging to the same retrotransposon lineages or clades occurring in different species. However, it should be noted that the taxon sampling was not even across all groups of seed plants, leaving some taxa poorly covered while some extensively studied families like Poaceae represented substantial portions of the sequence data. An additional limitation of our approach was imposed by the method employed for LTR retrotransposon identification that required a number of structural and sequence features to be preserved in element sequences in order to be detected. It is likely that these requirements biased the identification procedure towards recently active elements that have not yet accumulated mutations eventually obscuring these features. On the other hand, such approaches that mostly rely on structural features of LTR retrotransposons instead of their sequence similarities are well suited for the identification of novel elements and have been successfully used in a number of studies (McCarthy et al. 2002; Macas and Neumann 2007; Neumann et al. 2011). Moreover, high mutation frequency in older elements, should they be included in the analysis, would hamper eORF identification due to the occurrence of multiple frameshift and nonsense mutations.
A relatively large number of novel elements containing eORFs were identified, including representatives of all three major lineages of plant Ty3/gypsy retrotransposons. The highest proportion of elements carrying eORFs was found in the Tat lineage, which also exhibited remarkable variability in eORF sequences and in their localization within the elements. Sequence similarities detected between 5′ eORFs from the group A and 3′ eORF from the group B elements (Fig. 3) strongly suggest that eORFs in these two groups are of common origin. Since the elements of group B include both, dicot and monocot species as opposed to group A made of the distinct clade of Ogre-like elements confined to a subset of dicot taxa, it appears more likely that the 3′ located eORFs represent the ancestral type. This is supported by the observation of the same type of eORF arrangement in the remaining two groups (C and D) and by the occurrence of hyper-variable regions within 3′ UTRs of Tat elements (Macas et al. 2009) that may promote the integration of foreign sequences. On the other hand, the presence of a plant mobile domain (Babu et al. 2006) in A and B group eORFs favors their origin from an insertion event of a transposable element over the transduction of a gene fragment. There were also several exceptions from the sequence organization typical of group A found in elements that clearly belonged to this group according to phylogenetic analysis of their RT sequences (Fig. 3). They included Ogre-SL from Silene latifolia where the eORF was arranged as in the group B elements (Fig. 2). A similarity search using this eORF protein sequence was performed in order to identify additional Ogre-like elements with a sequence organization similar to Ogre-SL. This search yielded only one partial element that was identified in the genomic sequence (GenBank accession AP011970.1, position 1-3516) of Jatropha curcas (Euphorbiaceae). However, due to the lack of a RT domain, this element could not be included in the tree shown in Fig. 3. On the other hand, it was found that in the population of Ogre-like elements from Vitis vinifera showing high similarities of their RT domains to Ogre-SL (see the element Ogre-VV on Fig. 3) there were no eORFs detected.
A unique eORF organization was also found for GmOgre which contained a 5′ eORF sequence that was similar to those from other Ogre elements, while its 3′ eORF showed partial similarity to eORFs located at a similar position in elements from the Athila lineage (Du et al. 2010 and our results). In addition, the similarity of GmOgre 3′ eORF to Ty1/copia SIRE-like elements was detected by Laten et al. (2009) using PSI-BLAST, although this similarity appears to be limited to a few short regions when the direct comparison of predicted protein sequences is performed (data not shown). It seems that the Tat/Ogre-like elements containing this 3′ eORF are rather exceptional because all identified elements belonged to the same family as GmOgre and were detected in the Glycine max genome only. It is of interest that Athila-like (Calypso and Gm1) and SIRE-like elements are also present in the G. max genome, suggesting the possibility of insertion- or recombination-based capture of their sequences by the ancestral GmOgre elements.
The 3′ position and forward orientation of eORFs in the Athila lineage have previously led to the designation of this group as endogenous retroviruses, supposing that the eORFs represented envelope-like genes (Wright and Voytas 2002; Peterson-Burch et al. 2000). Since the retroviral env genes as well as Athila-like 3′ eORFs have heterogeneous sequences, this hypothesis was supported only indirectly by the common presence of transmembrane domains encoded by these sequences and by RNA splicing resembling that of genuine animal-infecting retroviruses. However, the transmembrane domains are ubiquitous across a broad spectrum of proteins (Krogh et al. 2001) and their experimental evidence in the putative Env-like proteins is missing. Some proteins encoded by 3′ eORFs even lack the predicted transmembrane domains (Fig. 4), and the 3′ eORFs themselves are lacking in a large part of the elements (Table 1 and Yano et al. 2005; Marín and Lloréns 2000). Moreover, the splicing observed in the BAGY-2 has not been experimentally demonstrated in other elements (Neumann et al. 2005). Taken together, the designation of the 3′ eORFs as putative envelope-like genes is questionable. This view is further supported by our finding that one group of elements acquired the eORF from a cellular gene (LOGL4) of known catalytic function, indicating that at least some elements from the Athila lineage possess eORF which clearly does not have the function of the retroviral Envelope protein. High sequence diversity of the eORFs also suggests that they were acquired independently from different sources and that their function, if any, differs between various representatives of the Athila lineage.
The smallest proportion of elements carrying eORF was detected in the lineage of Chromoviruses (Table 1) where eORFs were limited to 5′ regions of one group of phylogenetically related elements (Fig. 5). The lack of 3′ located eORFs in Chromoviruses is due to the specific structure of their gag-pol regions that are extended close to or within 3′ LTRs (Neumann et al. 2011). The occurrence of 5′ eORFs in elements from only one plant family (Poaceae) and their mutual similarities suggest a common origin.
Two fundamental questions arise with respect to the widespread occurrence of eORFs in plant Ty3/gypsy elements, the first one concerning their origin and the second regarding their expression and translation into proteins. Even the substantial volumes of genomic data available from a variety of plant species which were screened for similarities to the identified eORF sequences did not provide conclusive information about the origin of most of the eORFs. However, in the case of Tat group A and B eORFs containing plant mobile domains, it may be the case that they are derived from an insertional event involving DNA transposon (Babu et al. 2006), followed by degradation or deletion of most of the original transposon sequence. Transduction of cellular gene sequences which have been well documented for retroviruses (Coffin et al. 1997) represent another potential mechanism of eORF origin in Ty3/gypsy retrotransposons, as they are closely related to retroviruses. However, the only cases where a clear similarity pointing to such an event was found included the Athila-like elements Cs1 a Pt1 (similarity to LOGL4 gene; Kuroha et al. 2009) and the partial similarity to the histone H3 sequence found in some Chromoviruses (Figs. 5, 6). The latter case resembles the chimeric nature of the ORF1 in the Bs1 element which originated by the fusion of a transduced cellular gene sequences with a truncated retrotransposon ORF (Jin and Bennetzen 1994; Elrouby and Bureau 2001).
The question of whether eORFs can be expressed and translated into proteins is crucial for investigating their potential importance for their carriers. While the eORFs that are in the sense orientation are expressed as parts of native retrotransposon transcripts originating from promoters within the 5′ LTR and spanning the whole internal region (part of the 3′ LTR as well), it is not clear if and how they are translated into proteins. The 5′ eORFs of Tat/Ogre elements and Chromoviruses could actually be readily translated as they represent the first ORFs preceding the ones encoding Gag-Pol proteins (Fig. 2). However, translation of these proteins which are essential for retrotransposon replication would be disabled unless some additional mechanism that allows for the translation of the gag-pol could be employed. A similar problem applies to the translation of eORFs located downstream of the gag-pol region within polycistronic gag-pol/3′eORF transcripts. Several mechanisms of translational recoding have been described that could facilitate co-translation of separated ORFs from LTR retrotransposon transcripts by ribosomal frameshifting or stop codon bypass (Gao et al. 2003; Forbes et al. 2007). In addition, transcript splicing has been reported to have a role in the removal of intron sequences that are located within gag-pol regions of Ogre elements (Steinbauerová et al. 2008). The same mechanism was proposed to allow translation of 3′ eORFs of BAGY-2 (Athila lineage) by fusing it with the gag ORF (Vicient et al. 2001). In the case of eORFs in the opposite orientation relative to the gag-pol ORF, the generation of additional, antisense transcripts are required for their expression. Such antisense transcripts initiated in the 3′ LTR and allowing the expression of a protein encoded by reverse-oriented ORF were reported for the retroviruses HTLVs (Barbeau and Mesnard 2011). There are indications that antisense promoters exist also in plant LTR retrotransposons (Kato et al. 2005), however, it is yet to be investigated if this mechanism is common in the elements carrying eORFs.
References
Babu MM, Iyer LM, Balaji S, Aravind L (2006) The natural history of the WRKY-GCM1 zinc fingers and the relationship between transcription factors and transposons. Nucleic Acids Res 34:6505–6520
Barbeau B, Mesnard J-M (2011) Making sense out of antisense transcription in human T-cell lymphotropic viruses (HTLVs). Viruses 3:456–468
Benson G (1999) Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27:573–580
Coffin JM, Hughes SH, Varmus HE (1997) Retroviruses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Du J, Tian Z, Hans CS, Laten HM, Cannon SB, Jackson SA, Shoemaker RC, Ma J (2010) Evolutionary conservation, diversity and specificity of LTR-retrotransposons in flowering plants: insights from genome-wide analysis and multi-specific comparison. Plant J 63:584–598
Elrouby N, Bureau TE (2001) A novel hybrid open reading frame formed by multiple cellular gene transductions by a plant long terminal repeat retroelement. J Biol Chem 276:41963–41968
Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunasekaran P, Ceric G, Forslund K, Holm L, Sonnhammer EL, Eddy SR, Bateman A (2010) The Pfam protein families database. Nucleic Acids Res 38:D211–D222
Forbes EM, Nieduszynska SR, Brunton FK, Gibson J, Glover LA, Stansfield I (2007) Control of gag-pol gene expression in the Candida albicans retrotransposon Tca2. BMC Mol Biol 8:94
Gao X, Havecker ER, Baranov PV, Atkins JF, Voytas DF (2003) Translational recoding signals between gag and pol in diverse LTR retrotransposons. RNA 9:1422–1430
Gao D, Gill N, Kim H-R, Walling JG, Zhang W, Fan C, Yu Y, Ma J, SanMiguel P, Jiang N, Cheng Z, Wing RA, Jiang J, Jackson SA (2009) A lineage-specific centromere retrotransposon in Oryza brachyantha. Plant J 60:820–831
Gorinsek B, Gubensek F, Kordis D (2004) Evolutionary genomics of chromoviruses in eukaryotes. Mol Biol Evol 21:781–798
Havecker ER, Gao X, Voytas DF (2004) The diversity of LTR retrotransposons. Genome Biol 5:225
Hawkins JS, Grover CE, Wendel JF (2008) Repeated big bangs and the expanding universe: directionality in plant genome size evolution. Plant Sci 174:557–562
Hofmann K, Stoffel W (1993) TMBASE—a database of membrane spanning protein segments. Biol Chem H-S 374:166
Hu TT, Pattyn P, Bakker EG et al (2011) The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nat Genet 43:476–481
International Brachypodium Initiative (2010) Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 463:763–768
Jaillon O, Aury J-M, Noel B et al (2007) The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449:463–467
Jin YK, Bennetzen JL (1994) Integration and nonrandom mutation of a plasma membrane proton ATPase gene fragment within the Bs1 retroelement of maize. Plant Cell 6:1177–1186
Kato A, Endo M, Kato H, Saito T (2005) The antisense promoter of AtRE1, a retrotransposon in Arabidopsis thaliana, is activated in pollens and calluses. Plant Sci 168:981–986
Kejnovsky E, Kubat Z, Macas J, Hobza R, Mracek J, Vyskot B (2006) Retand: a novel family of gypsy-like retrotransposons harboring an amplified tandem repeat. Mol Genet Genomics 76:254–263
Krogh A, Larsson B, von Heijne G, Sonnhammer EL (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305:567–580
Kumar A, Bennetzen JL (1999) Plant retrotransposons. Annu Rev Genet 33:479–532
Kumekawa N, Ohtsubo H, Horiuchi T, Ohtsubo E (1999) Identification and characterization of novel retrotransposons of the gypsy type in rice. Mol Gen Genet 260:593–602
Kuroha T, Tokunaga H, Kojima M, Ueda N, Ishida T, Nagawa S, Fukuda H, Sugimoto K, Sakakibara H (2009) Functional analyses of LONELY GUY cytokinin-activating enzymes reveal the importance of the direct activation pathway in Arabidopsis. Plant Cell 21:3152–3169
Laten HM, Mogil LS, Wright LN (2009) A shotgun approach to discovering and reconstructing consensus retrotransposons ex novo from dense contigs of short sequences derived from Genbank Genome Survey Sequence database records. Gene 448:168–173
Li W, Jaroszewski L, Godzik A (2001) Clustering of highly homologous sequences to reduce the size of large protein databases. Bioinformatics 17:282–283
Li W, Jaroszewski L, Godzik A (2002) Tolerating some redundancy significantly speeds up clustering of large protein databases. Bioinformatics 18:77–82
Lloréns C, Futami R, Bezemer D, Moya A (2008) The gypsy database (GyDB) of mobile genetic elements. Nucleic Acids Res 36:D38–D46
Loidl P (2004) A plant dialect of the histone language. Trends Plant Sci 9:84–90
Macas J, Neumann P (2007) Ogre elements—a distinct group of plant Ty3/gypsy-like retrotransposons. Gene 390:108–116
Macas J, Koblížková A, Navrátilová A, Neumann P (2009) Hypervariable 3′ UTR region of plant LTR-retrotransposons as a source of novel satellite repeats. Gene 448:198–206
Macas J, Kejnovský E, Neumann P, Novák P, Koblížková A, Vyskot B (2011) Next generation sequencing-based analysis of repetitive DNA in the model dioecious plant Silene latifolia. PLoS ONE 6:e27335
Marchler-Bauer A, Lu S, Anderson JB et al (2011) CDD: a conserved domain database for the functional annotation of proteins. Nucleic Acids Res 39:D225–D229
Marín I, Lloréns C (2000) Ty3/Gypsy retrotransposons: description of new Arabidopsis thaliana elements and evolutionary perspectives derived from comparative genomic data. Mol Biol Evol 17:1040–1049
Martínez-Izquierdo JA, García-Martínez J, Vicient CM (1997) What makes Grande1 retrotransposon different? Genetica 100:15–28
McCarthy EM, Liu J, Lizhi G, McDonald JF (2002) Long terminal repeat retrotransposons of Oryza sativa. Genome Biol 3 (RESEARCH0053)
Ming R, Hou S, Feng Y et al (2008) The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus). Nature 452:991–996
Neumann P, Požárková D, Macas J (2003) Highly abundant pea LTR retrotransposon Ogre is constitutively transcribed and partially spliced. Plant Mol Biol 53:399–410
Neumann P, Požárková D, Koblížková A, Macas J (2005) PIGY, a new plant envelope-class LTR retrotransposon. Mol Genet Genomics 273:43–53
Neumann P, Koblížková A, Navrátilová A, Macas J (2006) Significant expansion of Vicia pannonica genome size mediated by amplification of a single type of giant retroelement. Genetics 173:1047–1056
Neumann P, Navrátilová A, Koblížková A, Kejnovský E, Hřibová E, Hobza R, Widmer A, Doležel J, Macas J (2011) Plant centromeric retrotransposons: a structural and cytogenetic perspective. Mobile DNA 2:4
Novák P, Neumann P, Macas J (2010) Graph-based clustering and characterization of repetitive sequences in next-generation sequencing data. BMC Bioinform 11:378
Ohtsubo H, Kumekawa N, Ohtsubo E (1999) RIRE2, a novel gypsy-type retrotransposon from rice. Genes Genet Syst 74:83–91
Ouyang S, Zhu W, Hamilton J et al (2007) The TIGR rice genome annotation resource: improvements and new features. Nucleic Acids Res 35:D883–D887
Paterson AH, Bowers JE, Bruggmann R et al (2009) The Sorghum bicolor genome and the diversification of grasses. Nature 457:551–556
Pearson WR, Lipman DJ (1988) Improved tools for biological sequence comparison. Proc Natl Acad Sci USA 85:2444–2448
Pearson WR, Wood T, Zhang Z, Miller W (1997) Comparison of DNA sequences with protein sequences. Genomics 46:24–36
Pereira V (2004) Insertion bias and purifying selection of retrotransposons in the Arabidopsis thaliana genome. Genome Biol 5:R79
Peterson-Burch BD, Wright DA, Laten HM, Voytas DF (2000) Retroviruses in plants? Trends Genet 16:151–152
Schmutz J, Cannon SB, Schlueter J et al (2010) Genome sequence of the palaeopolyploid soybean. Nature 463:178–183
Schnable PS, Ware D, Fulton RS et al (2009) The B73 maize genome: complexity, diversity, and dynamics. Science 326:1112–1115
Steinbauerová V, Neumann P, Macas J (2008) Experimental evidence for splicing of intron-containing transcripts of plant LTR retrotransposon Ogre. Mol Genet Genomics 280:427–436
Tuskan GA, Difazio S, Jansson S et al (2006) The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313:1596–1604
Vicient CM, Kalendar R, Schulman AH (2001) Envelope-class retrovirus-like elements are widespread, transcribed and spliced, and insertionally polymorphic in plants. Genome Res 11:2041–2049
Wicker T, Keller B (2007) Genome-wide comparative analysis of copia retrotransposons in Triticeae, rice, and Arabidopsis reveals conserved ancient evolutionary lineages and distinct dynamics of individual copia families. Genome Res 17:1072–1081
Wright DA, Voytas DF (2002) Athila4 of Arabidopsis and Calypso of soybean define a lineage of endogenous plant retroviruses. Genome Res 12:122–131
Xu Z, Wang H (2007) LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res 35:W265–W268
Yano ST, Panbehi B, Das A, Laten HM (2005) Diaspora, a large family of Ty3-gypsy retrotransposons in Glycine max, is an envelope-less member of an endogenous plant retrovirus lineage. BMC Evol Biol 5:30
Zuccolo A, Sebastian A, Talag J, Yu Y, Kim H, Collura K, Kudrna D, Wing RA (2007) Transposable element distribution, abundance and role in genome size variation in the genus Oryza. BMC Evol Biol 7:152
Acknowledgments
We thank Jasper E. Manning for his help with manuscript preparation. This work was supported by grants AVOZ50510513 from the Academy of Sciences of the Czech Republic, and P501/12/G090 from the Czech Science Foundation.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Steinbauerová, V., Neumann, P., Novák, P. et al. A widespread occurrence of extra open reading frames in plant Ty3/gypsy retrotransposons. Genetica 139, 1543–1555 (2011). https://doi.org/10.1007/s10709-012-9654-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10709-012-9654-9