
Abstract
Oxytropis chankaensis Jurtz. (Fabaceae) is an endangered perennial tetraploid species endemic to the Khanka Lake coast. In Russia, O. chankaensis is distributed across a very restricted zone along the western shore of this lake. To characterise all known populations of this species, we assessed the genetic diversity of four noncoding regions of chloroplast DNA (cpDNA). Variable sites detected within the trnL–trnF, the petG–trnP, and the trnS–trnG regions allowed the identification of seven haplotypes. On the other hand, no variation was found in the trnH–psbA region. O. chankaensis exhibited an overall low level of nucleotide diversity (π = 0.00052) but a marked haplotype diversity (h = 0.718). A combination of three or four haplotypes was found in each population, and most of the cpDNA variation (above 90%) was distributed within populations. The level of genetic structure that we detected in O. chankaensis using maternal plastid DNA markers was much lower (GST = 0.037) than the average that is estimated for angiosperms. We found no evidence for isolation by distance or for phylogeographic structuring in O. chankaensis. Our data suggest that autopolyploidy has arisen more than once in the evolutionary history of this species. Repetitive expansion and contraction during past and ongoing demographic events both seem to be involved in shaping the current genetic structure of O. chankaensis. This study provides valuable information for developing the most appropriate strategy for conserving this endemic species with a narrow habitat range.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Oxytropis (Fabaceae) is a taxonomically complex genus that comprises above 300 species (Zhu et al. 2010), of which 55 occur in the Russian Far East (Pavlova 1989). Among these, there are many rare and endemic species with restricted ranges. Oxytropis chankaensis Jurtz. (synonym O. hailarensis subsp. chankaensis (Jurtzev) Kitag.) is an endangered perennial herb with a narrow range that is restricted, in Russia, to the west shore of Khanka Lake (Kharkevich and Kachura 1981; Pavlova 1989; Yurtsev 1964), the largest lake in Northeast Asia (Fig. 1a). On the Chinese side of Khanka Lake, the only known occurrence of O. chankaensis is on a natural sandy spit lying between Khanka Lake and small Xiaoxingkai Lake (I.V. Maslova, the Researcher of the Khankaisky Nature Reserve, pers. com.). O. chankaensis is listed as “vulnerable” in the Rare Plant Species Book of the Far East of Russia (Kharkevich and Kachura 1981) and in the Red Data Book of the Primorsky Kray (2008). This legume was originally found on the shoreline of Khanka Lake and was first described as the distinct endemic species, Oxytropis chankaensis, based on definite morphological differences from its congeners (Yurtsev 1964). Later, the status of this species was reduced to a subspecies of O. hailarensis Kitag. (Kitagawa 1979), the species that occurs in China and Mongolia (Bisby et al. 2009; Czerepanov 1995). In the Flora of China, O. hailarensis is regarded as O. oxyphylla (Pall) DC (Zhu et al. 2010). However, the species from the west shore of Khanka Lake is clearly distinguishable from a related species, O. oxyphylla, not only by morphological features (Yurtsev 1964), but also by chromosome number: the latter species is diploid (2n = 16, Agapova et al. 1990; Zhu et al. 2010), while O. chankaensis is tetraploid (2n = 32, Gurzenkov and Pavlova 1984; Probatova et al. 2008). When we surveyed all of the present-day localities of O. chankaensis, we did not find any diploid or triploid plants. Hence, following Russian botanists who previously studied native plants of this species in the wild (Barkalov and Kharkevich 1996; Pavlova 1989; Probatova et al. 2008; Red Data Book Primorsky Kray 2008), we regard the Khanka Lake populations as a separate species, Oxytropis chankaensis Jurtz. belonging to subsect. Oxyphylliformes Jurtz. sect. Baicalia Steller ex Bunge subgenus Oxytropis.

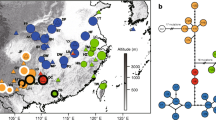
a Map of the southern Russian Far East showing the location of Khanka Lake. b Geographic location of the known localities of Oxytropis chankaensis on the west Russian shore of Khanka Lake (according to Kharkevich and Kachura 1981) and map of the frequencies of the seven chloroplast DNA (cpDNA) haplotypes observed in five populations of O. chankaensis. Filled circles with a code denote the sampling localities (for population codes, see Table 1). Open circles denote localities where O. chankaensis plants were found previously but are not found at present. Pie charts represent the proportion of haplotypes in each population. c Single most parsimonious tree from analysis of the chloroplast noncoding regions. The numbers above branches indicate bootstrap values based on 1,000 replicates. d The statistical parsimony (95%) network of cpDNA haplotypes constructed using TCS program. The size of circles corresponds to the frequency of each haplotype. Small open circle represents the inferred intermediate haplotype not detected in this study. Each line between haplotypes represents a mutational step. The H7 haplotype was identified by TCS as the ancestral haplotype
O. chankaensis is an outcrossing species that is pollinated by bumblebees, with pollen potentially being dispersed over long distances. Plants have high fecundity with fruits containing up to twenty seeds, and an individual plant produces approximately four thousand seeds (Kholina et al. 2003). Mature spherical pods can be dispersed by wind and water over long distances beyond the limits of local populations, while some seeds from the dehiscent pods are gravity-dispersed to only a short distance from the maternal plant to form the soil seed bank. O. chankaensis is a long-lived species with overlapping generations. O. chankaensis plants occur only in sandy habitats on a narrow strip along the west Khanka Lake coast, forming separate populations numbering from approximately 80 to 500 individuals (Kholina and Kholin 2006). Khanka Lake has always experienced variations in its shoreline, and the water level of Khanka Lake not only showed significant fluctuations during transgressions and regressions of previous epochs (Korotkii et al. 2007), but regular oscillations in the water level are also observed at present. Such oscillations in the water level and increasing anthropogenic effects may be the cause of the severe fluctuations in individual O. chankaensis population sizes that have been observed. In recent field surveys (2001–2005), we have not found this species in all localities where its presence was previously documented (Fig. 1b) and, in the localities where it is currently found, population sizes may vary from year to year from approximately twenty to several hundred plants (Kholina et al. 2009).
Knowledge of the population genetic structure of narrowly occurring endemic plants is of great importance for the purpose of conservation of the existing populations (Ellstrand and Elam 1993). Such information also provides an opportunity to gain a better understanding into how evolutionary processes such as speciation can act over small spatial scales (Prentice et al. 2003). Various factors (geographical range, taxonomic status, life form, breeding system, and dispersal capabilities) can influence the degree and distribution of genetic variation and gene flow within the range of a species (Hamrick and Godt 1996; Nybom 2004). Different historical processes also have influenced the spatial genetic structure of plant species. Assessing the contribution of each of these mechanisms to current population structure can prove difficult. The analysis of variation in plant genomes with different modes of inheritance can, however, provide valuable information on genetic structure of current populations and on evolutionary forces that have shaped the current distribution of genetic variation (Pleines et al. 2009 and references therein).
In earlier studies, we analysed intraspecific variation in O. chankaensis based on morphological features and on allozyme and random amplified polymorphic DNA (RAPD) markers that are assumed to represent the nuclear genome. Based on the results of canonical, discriminate and cluster analyses, distinct morphological differences between the populations were found (Kholina and Kholin 2008). High levels of genetic polymorphism and statistically significant differences among populations were also revealed by using both RAPD and allozyme markers (Artyukova et al. 2004; Kholina et al. 2007, 2009). In addition, allozyme analysis allowed us to identify southern and northern groups of populations that differed in the levels of allozyme diversity (Kholina et al. 2009). The tetrasomic segregation of the allozymes that we observed in O. chankaensis implies that this species is an autotetraploid (Kholina et al. 2004). The high levels of genetic and genotypic diversity that we found in this species based on allozyme data implied that polyploidy might have resulted from recurrent crosses between genetically different plants. Polyploidy is believed to be a common mechanism in the evolution of plants, and multiple origins have been shown for both auto- and allopolyploid plants including Oxytropis species (Jorgensen et al. 2003; Segraves et al. 1999; Soltis and Soltis 2000; Soltis et al. 2003, 2007; Tremetsberger et al. 2009). The polytopic and recurrent origin of many polyploids may account for the overall complexities that are observed in such taxa (Gauthier et al. 1997; Parisod et al. 2010; Soltis and Soltis 2009).
The combined use of nuclear and chloroplast DNA (cpDNA) data allows for the determination of the origin and evolutionary history of polyploids (Guo et al. 2006; Kao 2008; Soltis et al. 2003). CpDNA is predominantly transmitted through the seeds in most angiosperms including the Fabaceae (Doyle et al. 2004; Gauthier et al. 1997) and usually exhibits geographically structured variation (Korpelainen 2004; Petit et al. 2005). In recent decades, cpDNA markers have been widely used in investigations of genetic structure, phylogeography, and the reconstruction of the evolutionary history of endemic and endangered species (e.g., Artyukova et al. 2009; Ayele et al. 2009; Ikeda et al. 2008; Prentice et al. 2003; Wang et al. 2009). In the present study, the genetic structure of O. chankaensis was determined using sequence polymorphisms in four noncoding cpDNA regions. We addressed the following questions: (1) what level of plastid genome variability exists in this narrow-range endemic species; (2) how is genetic variation distributed within and among populations; and (3) what forces were involved in shaping the population structure.
Materials and methods
Plant materials
We studied individuals of O. chankaensis in the wild at localities where it had been reported in published data (Kharkevich and Kachura 1981; Pavlova 1989) and sampled the entire natural range of the species in Russia. Plants of O. chankaensis were only found in five out of eight previously documented localities (Fig. 1b). Three localities are located near the villages Turii Rog, Novokachalinsk, and Troitskoe, and two localities are situated in the territory of the Khankaiskii Nature Reserve. Specimens that are representative of the different ecotypes of O. chankaensis are held at the Herbarium of the Institute of Biology and Soil Science, Vladivostok (VLA). At the time of sampling, the estimated population sizes varied from 77 to 512 with an average of 49.8 adult generative plants in population. In each population, leaves from randomly selected adult generative plants approximately 200 m apart were collected in a way that was not damaging to the plants sampled. Sample size, population code, and the geographic coordinates for each population are given in Table 1.
DNA amplification and sequencing
Total genomic DNA was extracted as described in Artyukova et al. (2004). To investigate cpDNA variation, we tested six noncoding intergenic spacer regions of the chloroplast genome: the trnD GUC–trnT GGU, the trnH GUG–psbA, the petG–trnP, the rpoB–trnC, the trnL intron–trnLF intergenic spacer (trnL–F), and the trnS GCU–trnG UUC (trnS–G) that have been found to be polymorphic in some plant species (Desplanqe et al. 2000; Huang et al. 2002; Shaw et al. 2005). To amplify these regions, we used previously published primer pairs (Table 2) and thermocycling conditions (Shaw et al. 2005). We failed to amplify the trnD GUC–trnT GGU region, though all of the other cpDNA fragments were successfully amplified for the 63 plants tested. Sequencing of the PCR-amplified products was carried out in both directions under the sequencing conditions described by Shaw et al. (2005) using a BigDye terminator v. 3.1 sequencing standard kit (Applied Biosystems) with the same primer pairs that were used for amplification. In addition, internal primers were used for sequencing the trnS–G and the trnL–F regions (Table 2). Although amplification of the rpoB–trnC region was successful for all individuals tested, sequencing reactions with the primers that were used for amplification failed repeatedly; this region was therefore excluded from subsequent analysis. Sequences were analysed on an ABI 3130 genetic analyzer (Applied Biosystems, USA). Forward and reverse sequences were assembled using the Staden Package v. 1.4 (Bonfield et al. 1995) and aligned manually with the SeaView program (Galtier et al. 1996). DNA fragments that contained substitutions and/or microsatellite variants were retested (reamplified and resequenced) to verify that our results were repeatable.
Data analysis
Because cpDNA does not recombine and is, therefore, equivalent to a single locus, sequences for the four fragments that we investigated were combined to derive the haplotype of each individual. Two repeats (a dinucleotide AT-motif within the trnL UAA intron and a mononucleotide poly-T motif within the petG–trnP intergenic spacer) varied in length. These repeats were included in the data set because repeatability tests allowed us to exclude PCR errors. These repeat variations were treated as point mutations, interpreting each increase or decrease of a single repeated unit as a single mutational event (Simmons and Ochoterena 2000). Maximum parsimony analysis (with gaps coded as a fifth base or as characters in a separate presence/absence matrix) was performed in PAUP* v. 4.0b10 (Swofford 2003). To represent all possible alternative pathways between haplotypes within a single figure, we carried out a statistical parsimony analysis with a 95% confidence limit for parsimony using the TCS program and coding indels as a fifth state (Clement et al. 2000).
Most population genetic analyses were performed using Arlequin v. 3.11 (Excoffier et al. 2005) and DnaSP v. 4.5 (Rozas et al. 2003) software. We calculated numbers of haplotypes (nH) and values of haplotype diversity (h) and nucleotide diversity (π). To detect departures from the standard neutral model of evolution, we performed Tajima’s D (Tajima 1989) and Fu’s F S (Fu 1997) tests using Arlequin and Fu and Li’s (1993) D* and F* tests using DnaSP. These tests show different degrees of sensitivity to deviation from neutrality caused by demography or selection. Significance levels of these tests were assessed by generating 10,000 random samples and using model-based simulations (Excoffier et al. 2005). Positive values indicate that haplotype classes are evenly represented, whereas negative values result from the presence of a rare haplotype. Significant positive values of Tajima’s D, Fu and Li’s D* and F* tests and insignificant values of Fu’s F S test would suggest that either balancing selection or a reduction in population size had occurred, while significant values for only Fu’s F S suggest that population growth is occurring (Fu 1997). Nonsignificant test results can also be informative by comparing whether the values of several tests, together, are uniformly positive or negative (Fu 1997). D and F S tests tend to be negative if there is an excess of rare variants, which is an indication of genetic hitchhiking/selective sweep or population growth. An excess of common variants is expected to be caused by population subdivision, population size reduction, or balancing selection, such that the resulting values of these tests tend to be positive (Fu 1997; Fu and Li 1993; Tajima 1989; Wright and Gaut 2005). To test for population demographic changes, a mismatch pairwise distribution analysis (MDA) that linked the number of differences between haplotypes and haplotype frequency was also performed using Arlequin. We assessed the fit of the observed mismatch distributions to a model by coalescent simulation of 10,000 samples using the sum of squared deviations (SSD) between the observed and expected mismatch distributions and the raggedness index (r) as test statistics. The 95% confidence intervals of the demographic parameters were estimated with 10,000 replicates. Unimodal patterns with low and insignificant values of SSD and r are typical for expanding populations, while the distribution is multimodal in populations of constant size (Rogers and Harpending 1992). Population stability was also inferred if the 95% confidence intervals for two parameters, scaled mutation rates before (θ 0) and after (θ 1 ) growth, overlapped, even if the P value of the SSD was not significant (Schneider and Excoffier 1999).
An analysis of molecular variance (AMOVA; implemented in Arlequin) was performed to estimate the distribution of genetic variation within and between populations and the values of pairwise genetic distances (F ST) between populations. The significance of the variance components was determined with a permutation test (10,000 replicates). Thresholds of significance for pairwise F ST values were estimated using the Bonferroni correction for multiple tests. To calculate and compare the two differentiation indices N ST and G ST, we used Permut v. 2.0 software (available from http://www.pierroton.inra.fr/genetics/labo/Software) with a permutation test (10,000 permutations). G ST makes use of only haplotype frequencies, whereas N ST takes into account the similarities of haplotypes. A higher value for N ST than G ST would be indicative of phylogeographic structure (Petit et al. 2005; Pons and Petit 1996). We also calculated genetic differentiation standardised to the maximum level that could be obtained for the observed amount of genetic variation (G′ST), as proposed by Hedrick (2005). To test for a correlation between geographic and genetic distances, we performed a Mantel test using Arlequin software with the matrices of genetic differentiation defined as F ST or as the ratio of F ST/(1 – F ST) (Rousset 1997) and testing for significance with a permutation procedure (1,000 replicates). The ratio of gene flow via pollen and seeds was estimated following Ennos (1994) as r = {[(1/G STb−1) × (1 + F IS)]−2(1/G STm−1)}/(1/G STm−1), where G STm and G STb are the estimates of subdivision at maternally inherited markers and at nuclear (allozyme) markers, respectively; and F IS is the heterozygote deficit based on the previous allozyme data (Kholina et al. 2009).
Results
PCR products of the trnH–psbA, petG–trnP, trnS–G, and trnL–F regions were successfully amplified and sequenced for all O. chankaensis individuals tested. A total of 2,798 bp of aligned chloroplast sequences were obtained, comprising 420, 524, 1,158, and 696 bp for the trnH–psbA, petG–trnP, trnS–trnG, and trnL–F regions, respectively. A low number of polymorphic sites was detected: all individuals had identical sequences for the trnH–psbA region, while a single individual differed for a number of repetitive units of an AT-motif within the trnL–F region, and four variable characters were found within two other regions: one site in the trnS–G region and three sites in the petG–trnP (Table 3). Among the individuals we studied, seven haplotypes were recognised as combinations of the variable sites, of which three were informative for parsimony. Sequences of four cpDNA regions for each haplotype were deposited in EMBL/GenBank (see Table 2 for accession numbers). Maximum parsimony analysis yielded a single MP tree with a topology that was irrespective of the indel coding mode used and which was characterised by three clades with low bootstrap values (Fig. 1c). Low levels of diversity at the population level usually lead to a lack of phylogenetic resolution, and networks are considered the most appropriate way to represent all possible relationships within a species (Schaal et al. 2003). The network that we produced by the statistical parsimony method (Fig. 1d) had topological congruence with the MP tree and represented haplotype relationships with a single change in one base pair between any two haplotypes, except for H6, which differed from H4 in one repetitive unit of a dinucleotide repeat. Four haplotypes were frequently sampled (>5%) and the most frequent haplotype, H2, was found in 41.3% of the individuals in our samples, while the other three haplotypes (H1, H3, and H4) were found in 30.2, 15.9, and 7.9% of our specimens, respectively. Three haplotypes (H5, H6, and H7) were found only in single individuals, and H7 represented an intermediate haplotype between the two most frequent haplotypes, H2 and H1. The H2 haplotype was found in individuals from all localities, and the other frequent haplotypes also occurred in more than one population: haplotypes H1 and H3 were found in four populations, and the H4 haplotype was restricted to the populations TR and NK (Table 1; Fig. 1b).
Genetic diversity estimates at population and species levels are summarised in Table 4. Within all populations, the levels of nucleotide diversity were similarly low, and haplotype diversity was high. At the species level, the values of π and h were 0.00052 and 0.7179, respectively. Among populations, genetic differentiation was low (G ST = 0.037), and standardisation based on the maximum level that could be obtained for the observed amount of genetic variation increased the estimate of population differentiation only to a very small degree (G′ST = 0.146). The index of population structure, N ST, which additionally considers the similarities between haplotype sequences, was 0.099, and the difference between G ST and N ST was not significant (P > 0.05), indicating a lack of phylogeographic structure (Petit et al. 2005; Pons and Petit 1996). AMOVA demonstrated that the majority of molecular variation was found within populations, and less than 10% of the total genetic variance was among the populations (Φ ST = 0.09, P = 0.029; Table 5). When the samples were divided into two groups according to their location in the southern (PS, SI, and KR) or northern (TR and NK) regions of the study area, the majority of molecular variance was still observed among individuals within populations (87.23%, P = 0.027; Table 5). The small amounts of genetic variation that were attributable to differences between regions (9.81% of the total) and among populations within groups (2.95% of the total), were not significant (P = 0.10). Most values of F ST between pairs of populations were not significantly different from zero (P > 0.1; Table 6), and only the population NK was differentiated from all other populations (P < 0.05). However, after Bonferroni correction for multiple testing, the F ST-value between NK and TR was no longer significant, and a comparison between NK and KR became marginally significant (P = 0.006). The Mantel tests showed that there was no significant effect of isolation by distance because of the lack of correlation between the matrices of geographic distances and F ST or linearised F ST (r = 0.166, P = 0.225 and r = 0.178, P = 0.216, respectively). Only 2.75% of the genetic distance was explained by geographical distances between the populations. The ratio of gene flow via pollen and seeds was calculated to be 4.56, which is indicative of seed dispersal appears to comprise a main component of the total gene flow in this species.
Putative mechanisms that causing the observed pattern of cpDNA polymorphism could be inferred from the results of neutrality tests and MDA, though these inferences should be regarded with caution because different processes can produce the same patterns (Fu 1997; Rogers and Harpending 1992). Most tests of neutrality that were calculated for the entire data set or for the populations were statistically insignificant (P > 0.10; Table 7), indicating that populations are in mutation–drift equilibrium. However, the values of all neutrality tests except for the NK populations tended to be positive, suggesting the presence of haplotypes with intermediate frequencies. In contrast, the fact that all of the tests for population NK had negative values resulted from the presence of rare haplotypes in this population. The shapes of mismatch distributions were bimodal for most populations and the entire data set that indicated the presence of two frequent haplotypes (Fig. 2). A bimodal distribution was shown to be consistent with the distribution pattern that would be expected in populations whose size had either stayed constant or contracted (Harpending et al. 1998; Slatkin and Hudson 1991). However, the SSD and r-index values were insignificant in all cases (Table 7), which did not allow us to reject an expansion model completely. It should be noted that, for populations KR and SI, the 95% confidence intervals for θ 0 and θ 1 overlapped, which was inconsistent with the hypothesis of population growth (Schneider and Excoffier 1999). For other populations, and for the entire data set, the 95% confidence intervals for θ 0 and θ 1 did not overlap, implying that these populations could have undergone an expansion in the past. Only for population TR (Fig. 2) was the shape of mismatch distributions unimodal with the SSD and r-index values fitting an expansion model.

Mismatch distribution of pairwise nucleotide differences in Oxytropis chankaensis. The lines with filled squares show the observed distributions of pairwise nucleotide differences between haplotypes both within populations and for the entire data set. Lines with small dots represent the expected distributions fitted to the data under a model of population expansion. For population codes, see Table 1
Discussion
O. chankaensis is an endemic species with a very narrow geographic range and high habitat specificity. Small, isolated populations of such endemic species are prone to genetic drift and inbreeding, which can erode genetic diversity, reduce fitness, and threaten the long-term survival of populations, even in the absence of habitat destruction (Ellstrand and Elam 1993). Previously, we found higher levels of genetic diversity based on allozyme (He = 0.301, Kholina et al. 2009) and RAPD (He = 0.290, unpublished data) markers in O. chankaensis, compared with the average values for endemic plants (He, alloz = 0.076, Godt et al. 1996 and He, RAPD = 0.20, Nybom 2004). In the present study, based on cpDNA, we found low nucleotide diversity. Our finding of few polymorphisms within a total of 2,798 bp DNA region is consistent with the low mutation rate in the chloroplast genome that has been estimated for genus Oxytropis of 8.9 × 10−10 substitutions per site per year (Wojeiechowski 2005). Likewise, low nucleotide diversity in cpDNA has been found in other endemic species (e.g., Petunia exserta,π = 0.0007, Lorenz-Lemke et al. 2006; Hymenaea stigonocarpa, π = 0–0.0027, Ramos et al. 2007; Aconitum gimnandrum,π = 0–0.0050, Wang et al. 2009), and there is no cpDNA variation at all in nine populations of Heptacodium miconioides (Lu et al. 2006).
The most striking feature of O. chankaensis that was revealed in our investigation is its unexpectedly high cpDNA haplotype diversity (h = 0.7179) for a species that is confined to a very restricted zone (less than 0.5 km2). High levels of total haplotype diversity have been observed in some widespread (e.g., Acacia acuminata, h = 0.9196, Byrne et al. 2002) and endemic species with wider areas of distribution (e.g., Phyllodoce nipponica, h = 0.852, Ikeda and Setoguchi 2007; Petunia exserta, h = 0.657, Lorenz-Lemke et al. 2006; Hymenaea stigonocarpa, h = 0.804, Ramos et al. 2007; Aconitum gimnandrum, h = 0.739; Wang et al. 2009). In most angiosperms, populations often are fixed for single cpDNA haplotypes, and polymorphic populations possessing different haplotypes occur in potential contact zones of the different maternal lines or at sites of long-term persistence. In contrast, no populations of O. chankaensis that we sampled were fixed for a single haplotype, and all populations displayed high levels of haplotype diversity, sharing two or three common haplotypes (Fig. 1b). The uniform levels of diversity that we detected across the range of O. chankaensis might be due to these populations originating from a once continuous ancestral population, and the occurrence of several cpDNA haplotypes in populations could be explained by polymorphisms that were present in the putative ancestor. According to coalescence theory (Posada and Crandall 2001), the H7 haplotype may be a more ancient haplotype because it is found in a central position in the network (Fig. 1d). However, the most frequent haplotype H2 could also be considered an old haplotype because it exhibits three connections to other haplotypes, and it is found at a high frequency in our study populations. The occurrence of both these haplotypes in population PS agrees with our previous results based on allozyme data, suggesting that this population could be, putatively, the centre of the species formation (Kholina et al. 2009).
The presence of different cpDNA haplotypes within the extremely narrow geographic range of O. chankaensis could also be explained by the recurrent polyploidy events in the evolutionary history of this species. If tetraploid O. chankaensis originated once, then we would expect little if any cpDNA diversity. Multiple origins of polyploidy could increase amounts of variation in a species, also by adding different maternal lineages (Parisod et al. 2010; Soltis and Soltis 2009), and the number of different cpDNA haplotypes in a species indicates the maximum possible number of origins of polyploidy (Segraves et al. 1999). Our results allowed us to assume that the current populations of O. chankaensis have originated from at least three polyploidization events. During the cold and dry periods of the Holocene, when forest-steppe occupied the southern part of the Russian Far East, the steppe vegetation, which could have included a putative diploid progenitor of O. chankaensis, was more widespread throughout the Khanka plain than now (Bazarova et al. 2008; Mokhova et al. 2009). The new tetraploid species could have arisen by several polyploidy events within a genetically diverse diploid parent population of steppe species. The ancestral forms might have been lost to extinction during subsequent climatic changes and recurrent transgressions and regressions of Khanka Lake, while newly formed polyploids survived in a narrow coastal zone due to greater genetic flexibility of polyploids (Rausch and Morgan 2005) and higher productivity. In some groups of plants, including Oxytropis species (Jorgensen et al. 2003), the presence of mixed cytotypes within populations or different cytotypes at adjacent territories is relatively common. However, no diploid or triploid plants of relative congeners that could be progenitors of tetraploid O. chankaensis occur in close proximity to the current range of this species. Some diploid (2n = 16) species of sect. Baicalia occur in China, Mongolia and Korea (Zhu et al. 2010), e.g., O. oxyphylla, O. lanata, O. myriophylla, O. pumila, and O. ochrantha. In the sight of Jurtzev (1964), O. koreana (which is now regarded as a synonym of O. racemosa, Bisby et al. 2009; Zhu et al. 2010) is the closest extant relative of O. chankaensis. However, it is difficult to identify which of these species could be a putative ancestor of O. chankaensis or these closely relative species of sect. Baicalia were descended from a common progenitor.
Fluctuations in population size due to lake-level oscillations can result in a loss of genetic diversity due to population bottlenecks. However, severe or long-lasting population bottlenecks are considered likely to result in the extinction of some haplotypes, though all of the haplotypes we found in O. chankaensis are contiguous to one another across the entire genetic network (Fig. 1d). The overall pattern of the genetic variation in noncoding regions of cpDNA is in agreement with the expectations of the neutral equilibrium model of evolution, though selection cannot be excluded entirely. However, it is unlikely that selection would strongly affect noncoding regions of cpDNA. The results of neutrality tests and MDA for population NK are consistent with a loss of intermediate haplotypes and a recent increase in a small number of surviving haplotypes (Fig. 2; Table 7). For most other populations, these results are consistent with a recent contraction or population stability, though expansion cannot be ruled out completely because the signs of past expansions could have been erased due to repetitive habitat size fluctuations. The smallest MDA age expansion parameter (τ, Table 7) for population NK and its distance from population TR imply that population NK could be recolonising from the population TR, which is the only population containing both haplotypes that are frequent in NK and where the traces of recent population expansion were detected using MDA (Fig. 2). The occurrence of unique haplotypes in the population NK might have resulted from new mutations or rare seed dispersal events from the Chinese part of species area where some O. chankaensis plants have been observed on a natural sandy spit lying between Khanka Lake and Xiaoxingkai Lake (I.V. Maslova, the Researcher of the Khankaisky Nature Reserve, pers. com.). The long genesis of Khanka Lake (presumably from the middle Miocene) is thought to have gone through a series of transgressions and regressions including nearly complete exsiccation and draining of the surrounding plains (Korotkii et al. 2007). At present Khanka Lake also has no constant shoreline, and the demography of O. chankaensis is characterised by potential instability through periodic habitat reduction and expansion events. Additionally, populations of O. chankaensis have become more fragmented due to anthropogenic influences in the region, which have increased during the last decades.
The variation of haplotype frequency among populations and the low, mostly insignificant, pairwise F ST values between them (Table 6) might be a consequence of genetic drift occurring after splitting of a formerly more continuous population. Spatial genetic patterning in cpDNA was determined to be very weak, and the standardised differentiation estimate for the plastid genome (G′ST = 0.146) is much lower than the average value that has been found for angiosperms (G ST = 0.637, Petit et al. 2005). In the majority of angiosperms, including endemic and polyploid species, plastid DNA is generally more highly structured compared with the nuclear genome (Kao 2008; Korpelainen 2004; Petit et al. 2005). In contrast, the low level of cpDNA subdivision across O. chankaensis populations (G ST = 0.037; Φ ST = 0.090) is comparable with the low levels of differentiation for the nuclear genome that were found previously from allozyme and RAPD markers (Φ ST-RAPD = 0.13, Kholina et al. 2007; G STalloz = 0.028, Kholina et al. 2009). Mantel test results for the plastid genome point to a lack of isolation by distance that agrees with the results based on allozyme and RAPD data (unpublished data). Low population partitioning (cohesive genetic system) and the lack of phylogeographic patterning (as judged from N ST/G ST comparison) may be attributed to both recent fragmentations and extensive gene flow. The ratio of gene flow via pollen and seeds that we estimated is low (4.56) as compared with the median value of 17 that was determined for 93 comparison species (Petit et al. 2005). This ratio is consistent with a hypothesis of interpopulation gene flow predominantly due to seed dispersal, which, in O. chankaensis, occurs mainly through dispersal of mature pods by wind and water, though random dispersal of seeds between populations by humans also cannot be ruled out.
Thus, the genetic structure that we observed in O. chankaensis can apparently be explained due to both biological traits and historical factors. Although our data do not allow us to make a conclusion about the origin of O. chankaensis with confidence, based on independent lines of evidence (data from noncoding region sequences of cpDNA, RAPDs, and allozymes; Artyukova et al. 2004; Kholina et al. 2004, 2007, 2009), the most likely hypothesis is that this species originated from a putative diploid progenitor by recurrent polyploidy events. The life history of O. chankaensis includes frequent extinction, recolonisation, and expansion due to repetitive fluctuations in the Khanka Lake water level. In spite of the capacity for rapid population growth through vigorous seed production (Kholina et al. 2003), the extinction of at least three local populations (Fig. 1b) occurring over the past 30–50 years is an obvious indication of the ongoing decline of O. chankaensis populations in number and in population size. The ultimate goals of conservation are to ensure the continuous survival of populations and to maintain their evolutionary potential. Our data show that the full complement of cpDNA haplotypes can be detected from three populations (PS, NK, and TR). However, the allozyme diversity data (Kholina et al. 2009) indicate that ensuring the presence of the full complement of alleles requires the inclusion of all populations. Given the potential instability of demography in this endemic species and its limited habitat preferences, we suggest conserving all five of the remnant populations, and the most suitable strategy for the conservation of O. chankaensis is the protection of its natural habitats.
References
Agapova ND, Arharova KB, Vahtina LI et al (1990) Chromosome numbers of flowering plants of the flora of the USSR: families Aceraceae–Menyanthaceae. In: Takhtajan A (ed) Numeri Chromosomatum Magnoliophytorum florae USSR. Nauka, Leningrad, pp 398–410
Artyukova EV, Kholina AB, Kozyrenko MM, Zhuravlev YN (2004) Analysis of genetic variation in rare endemic species Oxytropis chankaensis Jurtz. (Fabaceae) using RAPD markers. Russ J Genet 40:710–716
Artyukova EV, Kozyrenko MM, Gorovoy PG, Zhuravlev YN (2009) Plastid DNA variation in highly fragmented populations of Microbiota decussata Kom. (Cupressaceae), an endemic to Sikhote Alin Mountains. Genetica 137:201–212
Ayele TB, Gailing O, Umer M, Finkeldey R (2009) Chloroplast DNA haplotype diversity and postglacial recolonization of Hagenia abyssinica (Bruce) J.F. Gmel. in Ethiopia. Plant Syst Evol 280:175–185
Barkalov VY, Kharkevich SS (1996) The vascular plants of the Khankaysky reservation. Bot Zhurn (Leningrad) 81:104–116
Bazarova VB, Mokhova LM, Orlova LA, Belyanin PS (2008) Variation of the Lake Khanka level in the late Holocene, Primorye. Russ J Pac Geol 2:272–276
Bisby FA, Roskov YR, Orrell TM et al., (eds) (2009) Species 2000 & ITIS Catalogue of Life: 2009 Annual checklist. Digital resource at www.catalogueoflife.org/annual-checklist/2009/. Species 2000: reading, UK
Bonfield JK, Smith KF, Staden R (1995) A new DNA sequence assembly program. Nucleic Acids Res 23:4992–4999
Byrne M, Macdonald B, Coates D (2002) Phylogeographical patterns in chloroplast DNA variation within the Acacia acuminata (Leguminosae: Mimosoideae) complex in Western Australia. J Evol Biol 15:576–587
Clement M, Posada D, Crandall K (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1660
Czerepanov SK (1995) Vascular plants of Russia and adjacent states (the former USSR). Cambridge University Press, New York
Desplanqe B, Viard F, Bernard J et al (2000) The linkage disequilibrium between chloroplast DNA and mitochondrial DNA haplotypes in Beta vulgaris ssp. maritima (L.): the usefulness of both genomes for population genetic studies. Mol Ecol 9:141–154
Doyle JJ, Doyle JL, Rauscher JT, Brown AHD (2004) Diploid and polyploid reticulate evolution throughout the history of the perennial soybeans (Glycine subgenus Glycine). New Phytol 161:121–132
Ellstrand NC, Elam DR (1993) Population genetic consequences of small population size: implication for plant conservation. Ann Rev Ecol Syst 24:217–242
Ennos RA (1994) Estimating the relative rates of pollen and seed migration among plant populations. Heredity 72:250–259
Excoffier L, Laval G, Schneider S (2005) Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol. Bioinform Online 1:47–50
Fu Y-X (1997) Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147:915–925
Fu Y-X, Li W-H (1993) Statistical tests of neutrality of mutations. Genetics 133:693–709
Galtier N, Gouy M, Gautier C (1996) Seaview and phylo-win: two graphic tools for sequence alignment and molecular phylogeny. Comput Appl Biosci 12:543–548
Gauthier P, Lumaret R, Bedecarrats A (1997) Chloroplast-DNA variation in the genus Lotus (Fabaceae) and further evidence regarding the maternal parentage of Lotus corniculatus L. Theor Appl Genet 95:629–636
Godt MJW, Johnson BR, Hamrick JL (1996) Genetic diversity and population size in four rare southern Appalachian plant species. Conserv Biol 10:796–805
Guo Y-P, Vogl C, Van Loo M, Ehrendorfer F (2006) Hybrid origin and differentiation of two tetraploid Achillea species in East Asia: molecular, morphological and ecogeographical evidence. Mol Ecol 15:133–144
Gurzenkov NN, Pavlova NS (1984) Chromosome numbers of the representatives of the genera Astragalus and Oxytropis (Fabaceae) from the Far East of the USSR. Bot Zhurn (Leningrad) 69:1569–1570
Hamrick JL, Godt MJW (1996) Effects of life history traits on genetic diversity in plant species. Philos Trans R Soc Lond B Biol Sci 351:1291–1298
Harpending HC, Batzer MA, Gruven MA et al (1998) Genetic traces of ancient demography. Proc Natl Acad Sci USA 95:1961–1967
Hedrick PW (2005) A standardized genetic differentiation measure. Evolution 59(8):1633–1638
Huang SSF, Hwang S-Y, Lin T-P (2002) Spatial pattern of chloroplast DNA variation of Cyclobalanopsis glauca in Taiwan and East Asia. Mol Ecol 11:2349–2358
Ikeda H, Setoguchi H (2007) Phylogeography and refugia of the Japanese endemic alpine plant Phyllodoce nipponica Makino (Ericaceae). J Biogeogr 34:169–176
Ikeda H, Senni K, Fujuu N, Setoguchi H (2008) Consistent geographic structure among multiple nuclear sequences and cpDNA polymorphisms of Cardamine nipponica Franch. et Savat, (Brassucaceae). Mol Ecol 17:3178–3188
Jorgensen JL, Stehlik I, Brochmann C, Conti E (2003) Implication of ITS sequences and RAPD markers for the taxonomy and biogeography of the Oxytropis campestris and O. arctica (Fabaceae) complexes in Alaska. Am J Bot 90:1470–1480
Kao RH (2008) Origins and widespread distribution of co-existing polyploids in Arnica cordifolia (Asteraceae). Ann Bot 101:145–152
Kharkevich SS, Kachura NN (1981) Rare plant species of the soviet Far East and their conservation. Nauka, Moscow, pp 156–161
Kholina AB, Kholin SK (2006) Population age structure of rare plant Oxytropis chankaensis. In: Problems of preservation of wetlands of international meaning: Khanka Lake: Proceedings of the 2 Inter. Conf. Idea, Vladivostok, pp 26–35
Kholina AB, Kholin SK (2008) Intraspecific variation of Oxytropis chankaensis, Far Eastern endemic. Russ J Ecol 39:14–20. doi:10.1007/s11184-008-1003-y
Kholina AB, Markelova OV, Kholin SK (2003) Population structure and reproduction biology of the rare endemic species Oxytropis chankaensis Jurtz. In: Botanical researches in Asian Russia: proceedings of the XI congress of the Russian Botanical Society (18–22 August 2003, Novosibirsk–Barnaul), vol 3. Azbuka, Barnaul, pp 369–370
Kholina AB, Koren OG, Zhuravlev YN (2004) High polymorphism and autotetraploid origin of the rare endemic species Oxytropis chankaensis Jurtz. (Fabaceae) inferred from allozyme data. Russ J Genet 40:393–400
Kholina AB, Koren OG, Artyukova EV et al. (2007) High level of polymorphism in a narrow endemic Oxytropis chankaensis (Fabaceae) inferred from molecular markers. In: Kartavtsev YP and Kryukov AP (eds) Modern achievements in population, evolutionary and ecological genetics (MAPEEG–2007) Reg Found Dev Gen, Vladivostok, p 19
Kholina AB, Koren OG, Zhuravlev YN (2009) Genetic structure and differentiation of populations of the tetraploid species Oxytropis chankaensis (Fabaceae). Russ J Genet 45:70–80
Kitagawa M (1979) Neo-Lineamenta florae Manshuricae. J Cramer, Vaduz, pp 409–410
Korotkii AM, Grebennikova TA, Karaulova LP, Belyanina NI (2007) Lacustrine transgressions in the late Cenozoic Ussuri-Khanka depression (Primor’e). Russ J Pac Geol 1:352–365
Korpelainen H (2004) The evolutionary processes of mitochondrial and chloroplast genomes differ from those of nuclear genomes. Naturwissenschaften 91:505–518
Lorenz-Lemke AP, Mäder G, Muschner VC, Stehmann JR et al (2006) Diversity and natural hybridization in a highly endemic species of Petunia (Solanaceae): a molecular and ecological analysis. Mol Ecol 15:4487–4497
Lu H-P, Cai Y-W, Chen X-Y et al (2006) High RAPD but no cpDNA sequence variation in the endemic and endangered plant, Heptacodium miconioides Rehd. (Caprifoliaceae). Genetica 128:409–417
Mokhova L, Tarasov P, Bazarova V, Klimin M (2009) Quantitative biome reconstruction using modern and late quaternary pollen data from the southern part of the Russian Far East. Quat Sci Rev 28:2913–2926
Nybom H (2004) Comparison of different nuclear DNA markers for estimating intraspecific genetic diversity in plants. Mol Ecol 137:1143–1155
Parisod C, Holderegger R, Brochmann C (2010) Evolutionary consequences of autopolyploidy. New Phytol 186:5–17
Pavlova NS (1989) Family Fabaceae. In: Kharkevich SS (ed) The vascular plants of the Soviet Far East, vol 4. Nauka, Leningrad, pp 191–339
Petit RJ, Duminil E, Fineschi S et al (2005) Comparative organization of chloroplast, mitochondrial and nuclear diversity in plant populations. Mol Ecol 14:689–701
Pleines T, Jakob SS, Blattner FR (2009) Application of non-coding DNA regions in intraspecific analyses. Plant Syst Evol 282:281–294
Pons O, Petit RJ (1996) Measuring and testing genetic differentiation with ordered versus unordered alleles. Genetics 144:1237–1245
Posada D, Crandall KA (2001) Intraspecific gene genealogies: trees grafting into networks. Trends Ecol Evol 16:37–45
Prentice HC, Malm JU, Mateu-Andres I, Segarra-Moragues JG (2003) Allozyme and chloroplast DNA variation in island and mainland populations of the rare Spanish endemic, Silene hifacensis (Caryophyllaceae). Conserv Genet 4:543–555
Probatova NS, Seledets VP, Rudyka EG (2008) Oxytropis chankaensis. In: Marhold K. (ed.), IAPT/IOPB chromosome data 5. Taxon 57:560, E20
Ramos ACS, Lemos-Filho JP, Ribeiro RA et al (2007) Phylogeography of the tree Hymenaea stigonocarpa (Fabaceae: Caesalpinioideae) and the influence of quaternary climate changes in the Brazilian Cerrado. Ann Bot 100:1219–1228
Rausch JH, Morgan MT (2005) Effect of salf-fertilizatiom, inbreeding depression, and population size on autopolyploid establishment. Evolution 59:1867–1875
Red Data Book Primorsky Kray (2008) Plants. Rare and endangered species of plants and fungi. In: Kozhevnikov AE, Nedoluzhko VA, Barkalov VYu et al. (eds) AVK Apelsin, Vladivostok, pp 339–341
Rogers AR, Harpending H (1992) Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol 9:552–569
Rousset F (1997) Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145:1219–1228
Rozas J, Sanchez-DelBarrio JC, Messequer X, Rozas R (2003) DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19:2496–2497
Schaal BA, Gaskin JF, Caicedo AL (2003) Phylogeography, haplotype trees, and invasive plant species. J Hered 94:197–204
Schneider S, Excoffier L (1999) Estimation of past demographic parameters from the distribution of pairwise differences when the mutation rates vary among sites: application to human mitochondrial DNA. Genetics 152:1079–1089
Segraves KA, Thompson JN, Soltis PS, Soltis DE (1999) Multiple origins of polyploidy and the geographic structure of Heuchera grossulariifolia. Mol Ecol 8:253–262
Shaw J, Lickey EB, Beck JT et al (2005) The tortoise and the hare II: relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. Am J Bot 92:142–166
Simmons MP, Ochoterena H (2000) Gaps as characters in sequence-based phylogenetic analyses. Syst Biol 49:369–381
Slatkin M, Hudson RR (1991) Pairwise comparisons of mitochondrial DNA sequences in stable and exponentially growing populations. Genetics 129:555–562
Soltis PS, Soltis DE (2000) The role of genetic and genomic attributes in the success of polyploids. Proc Natl Acad Sci USA 97:7051–7057
Soltis PS, Soltis DE (2009) The role of hybridization in plant speciation. Annu Rev Plant Biol 60:561–588
Soltis DE, Soltis PS, Tate JA (2003) Advances in the study of polyploidy since plant speciation. New Phytol 161:173–191
Soltis DE, Soltis PS, Schemske DW et al (2007) Autopolyploidy in angiosperms: have we grossly underestimated the number of species? Taxon 56:13–30
Swofford DL (2003) PAUP*. Phylogenetic analysis using parsimony (*and other methods). Version 4. Sinauer Associates, Sunderland, Massachusetts
Taberlet P, Gielly L, Pautou G, Bouvet J (1991) Universal primers for amplification of three non-coding regions of chloroplast DNA. Plant Mol Biol 17:1105–1109
Tajima F (1989) The effect of change in population size on DNA polymorphism. Genetics 123:597–601
Tremetsberger K, UrtubeyY E, Terrab A (2009) Pleistocene refugia and polytopic replacement of diploids by tetraploids in the Patagonian and Subantarctic plant Hypochaeris incana (Asteraceae). Mol Ecol 18:3668–3682
Wang L, Abbott RJ, Zheng W et al (2009) History and evolution of alpine plants endemic to the Qinghai-Tibetan Plateu: Aconitum gimnandrum (Ranunculaceae). Mol Ecol 18:709–721
Wojeiechowski MF (2005) Astragalus (Fabaceae): a molecular phylogenetic perspective. Brittonia 57:382–396
Wright SI, Gaut BS (2005) Molecular population genetics and the search for adaptive evolution in plants. Mol Biol Evol 22:506–519
Yurtsev BA (1964) Conspectus of the system of section Baicalia Bge., genus Oxytropis DC. In: Linchevskii IA (ed) News in the systematics of higher plants. Nauka, Moscow-Leningrad, pp 191–218
Zhu X, Welsh SL., Ohashi H (2010) Oxytropis. In: Wu Z-Y, Raven PH, Hong DY (eds) Flora of China, vol 10, pp 453–500. Published on the Internet http://www.efloras.org(accessed 31 January 2010)
Acknowledgments
This work was supported by Grant of Russian Academy of Sciences № 09-I-P23-06 and partly by the Program “Molecular and Cell Biology” of the Presidium of the Russian Academy of Sciences (project no. 09-1-P22-03). The authors thank two anonymous reviewers for their helpful comments on the paper.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Artyukova, E.V., Kozyrenko, M.M., Kholina, A.B. et al. High chloroplast haplotype diversity in the endemic legume Oxytropis chankaensis may result from independent polyploidization events. Genetica 139, 221–232 (2011). https://doi.org/10.1007/s10709-010-9539-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10709-010-9539-8