
Abstract
We investigated genetic diversity of the hellbender (Cryptobranchus alleganiensis) throughout its range in the eastern US using nuclear markers and compared our results to a previously published mitochondrial analysis. A variety of nuclear markers, including protein-coding gene introns and microsatellites were tested but only microsatellites were variable enough for population level analysis. Microsatellite loci showed moderate among population sharing of alleles, in contrast to the reciprocal monophyly exhibited by mitochondrial DNA. However, analyses using F-statistics and Bayesian clustering algorithms showed considerable population subdivision and clustered hellbender populations into the same major groups as the mtDNA. The microsatellites combined with the mtDNA data suggest that gene flow is severely restricted or non-existent among eight major groups, and potentially among populations (rivers) within groups. The combined mtDNA and microsatellite data suggest that the currently recognized hellbender subspecies are paraphyletic. We suggest that the eight independent groups identified in our study should be managed as such, rather than basing conservation decisions on the two named subspecies of hellbender.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Genetic diversity should be incorporated into conservation planning (Moritz 2002; DeSalle and Amato 2004; Schwartz et al. 2007). As species become increasingly fragmented into small isolated populations, heterozygosity is lost and gene diversity is redistributed into between-population genetic variance (Templeton et al. 1990). If population sizes are small, isolated population fragments lose genetic variation and have an increased risk of extinction through inbreeding depression and reduced capacity to respond to selection (Templeton et al. 1990; Reed and Frankham 2003). If the population sizes of isolates are not too small they are likely to possess unique local adaptations acquired through selection. By treating all populations as genetically equivalent, management resources could inadvertently be diverted away from genetically unique populations. Further, by managing genetically distinct populations as genetically exchangeable, breeding programs or translocations could be detrimental by lowering population mean fitness via outbreeding depression or moving individuals to habitats to which they are not adapted (Gharrett et al. 1999; Hedrick 2001; Lenormand 2002). Using phylogeography to understand the distribution of genetic variation within and among geographically isolated populations will help to determine conservation priorities and management strategies (Beebee 2005).
Mitochondrial DNA (mtDNA) is frequently used as a marker for phylogeographic studies because of the ease of data collection, lack of recombination, faster rates of evolution (as compared to nuclear DNA), and effective haploidy. The latter means that reciprocal monophyly evolves more quickly for mtDNA than for nuclear genes (Moore 1995; Edwards and Beerli 2000; Hudson and Turelli 2003). However, mitochondrial gene trees are often criticized as not being representative of the actual species tree because of problems associated with random lineage sorting, introgression, and sex-biased gene flow (Moore 1995). Commonly, morphological, mitochondrial, and nuclear gene trees are discordant (Chan and Levin 2005). Thus, analysis of multiple independent loci can be more informative than single gene trees.
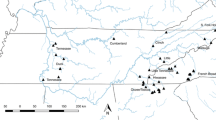
Worldwide, amphibians have been declining at a faster rate than other vertebrate taxa (Houlahan et al. 2000; Stuart et al. 2004). One of these declining amphibians, the hellbender, (Cryptobranchus alleganiensis) is among the largest of the North American salamanders (up to 74 cm in length, Nickerson and Mays 1973). Hellbenders are completely aquatic throughout their 30+ year lifespan (Nickerson and Mays 1973) and inhabit clear rocky fast-flowing streams in the eastern US (Fig. 1). Eastern hellbenders (C. a. alleganiensis) are found throughout the Appalachian Mountains from southern New York to northern Georgia and in rivers draining northward from the Ozarks. A second subspecies, the Ozark hellbender (C. a. bishopi), inhabits streams that drain south from the Ozark Plateau in Missouri and Arkansas and differs from the nominal subspecies in minor morphological characters (Grobman 1943). Because hellbenders breathe primarily through the skin, they are dependent on cool, well-oxygenated, flowing water (Guimond and Hutchison 1973). This specialized habitat requirements make hellbenders extremely vulnerable to the effects of habitat destruction including damming, increased sedimentation of rivers due to land development, and pollution from agricultural run-off (Williams et al. 1981; Mayasich et al. 2003; Phillips and Humphries 2005). Hellbender populations are also at risk from over-harvesting (Nickerson and Briggler 2007) and infection by chytrid fungus (Briggler et al. 2007). Recent studies have concluded that many populations of both subspecies are experiencing population declines (Williams et al. 1981; Wheeler et al. 2003, Routman, personal observation of the Meramec River population), and the US Fish and Wildlife Service (USFWS) is considering listing both subspecies as endangered under the Endangered Species Act (Amy Salveter, USFWS, pers. comm.).

Map of the eastern USA showing the hellbender sampling locations (modified from Sabatino and Routman 2009). Rivers where hellbenders were sampled are abbreviated as follows: Gasconade River (GR), Big Piney River (BP), Niangua River (NR), Meramec River (MR), Little River (LR), Beaverdam Creek (BC), Copper Creek (CC), Blue River (BR), Wabash River (WR), French Creek (FC), Slippery Rock Creek (SRC), New River (NR), West Fork of the Greenbrier River (WFGR), Sherman Creek (SC), North Fork of the White River (NFW), Spring River (SR), Current River (CR), Eleven Point River (EPR). The last four populations represent the subspecies C. a. bishopi. All other populations represent C. a. alleganiensis. Numbers on USA map are latitude and longitude scales
A previous allozyme study by Merkle et al. (1977) showed low within- and between-population genetic variation even between the two subspecies. They analyzed 24 loci from 12 populations sampled throughout the hellbender’s range. Twenty-two of these loci were monomorphic in all populations. The two remaining loci were polymorphic, but only in one population each. More recently, Routman et al. (1994) and Sabatino and Routman (2009) used mitochondrial DNA to gain insights into the population structure of hellbenders. Routman et al. (1994) found high levels of among-population variation using restriction enzyme digests of the entire mtDNA genome. Sabatino and Routman (2009) conducted a DNA sequence analysis using three mitochondrial genes. They determined hellbender populations were highly divergent and identified eight reciprocally monophyletic management units (MU). According to both Routman et al. (1994) and Sabatino and Routman (2009), the two named hellbender subspecies are paraphyletic if the mtDNA gene tree accurately reflects the population phylogeny.
In this study, we examine discrepancies between the morphological basis of the recognized subspecies and past genetic studies of C. alleganiensis with multiple independent nuclear markers. Nuclear loci in combination with the mitochondrial data will help evaluate the monophyly of the currently named hellbender subspecies and the existence of evolutionarily significant units (ESU) or MU. Furthermore, nuclear markers will suggest which populations, if any, are suitable for interbreeding or translocations, and which populations should be maintained as separate entities.
Materials and methods
Sampling
We obtained samples from 18 different rivers and streams throughout the current distribution of the hellbender, including populations from both subspecies. Blood and/or tissue samples used in this study were from previously collected samples (Fig. 1 and Table 1; see Routman 1993; Routman et al. 1994; Sabatino and Routman 2009) with the exception of six samples that were collected for this study from the West Fork of the Greenbrier River, a tributary of the New River in West Virginia. Sequencing of the mitochondrial cytochrome oxidase subunit I (COI) gene shows that these additional samples have unique mtDNA that is most closely related to the haplotypes from our earlier New River collection (data not shown).
Genetic data
For the previously collected samples, genomic DNA was extracted from blood or tissue using a standard phenol/chloroform method as outlined in Routman (1993). For the West Fork of the Greenbrier River samples, we extracted genomic DNA using a high salt extraction method similar to Aljanabi and Martinez (1997). All DNA samples were stored at −20°C.
A total of eight nuclear introns were surveyed for DNA sequence variation in a subsample of hellbenders from different populations (Appendix 1). Variation in these loci was minimal (Appendix 2), and the loci were not used for further analysis.
Four microsatellite loci developed by Johnson et al. (2009) were also analyzed: three dinucleotide microsatellite loci (CRAL1, CRAL4 and CRAL9) and one tetranucleotide locus (CRAL17). Microsatellite loci were amplified using fluorescent polymerase chain reaction (PCR) as described by Johnson et al. (2009). The forward primer was 5′-labelled with a florescent tag and the fluorescent product was then genotyped on an ABI 3100 sequencing machine (Applied Biosystems) and visualized using GeneScan Analysis Software (version 3.1; Applied Biosystems) with Filter Set D. The microsatellite loci exhibited a great deal of allelic diversity, and the analysis that follows is based solely on these loci.
Data analysis
To estimate the frequency of null alleles and scoring errors due to stuttering in our dataset we used micro-checker version 2.2.3 (Van Oosterhout et al. 2004). Several genetic statistics were calculated on the populations (number of alleles (Na), number of private alleles, and overall heterozygosity (H)) using the program GenAlEx version 6.1 (Peakall and Smouse 2006). We used arlequin version 3.11 (Schneider et al. 2000) to estimate observed (HO) and expected (HE) heterozygosities at each site and locus for statistically significant deviations from Hardy–Weinberg equilibrium. Significance was evaluated using MCMC (Markov chain parameters: 100,000 dememorization; 1,000,000 steps per chain) and we applied a Bonferroni correction (adjusted P value < 0.0009, α = 0.05). To test for linkage disequilibrium, we used GENEPOP version 4 (Raymond and Rousset 1995) with a Bonferroni correction (adjusted P value <0.0008, α = 0.05).
We used Structure version 2.2 (Pritchard et al. 2000) to infer population structure and assign individuals to genetic clusters. This software uses a Bayesian algorithm that creates clusters maximizing both Hardy–Weinberg equilibrium and gametic phase equilibrium within clusters and disequilibrium between clusters. Ten iterations were run for each value of K = 1 (rivers constitute a single panmictic population) through 20 (two more than the number of rivers sampled) we ran the independent model, with a burn-in of 50,000 and MCMC values of 500,000. We used the method described by Evanno et al. (2005) to identify the number of hierarchical clusters in our dataset. Clumpp 1.1.1 (Jakobsson and Rosenberg 2007) was used to average the results of multiple iterations for a given K value. The program Distruct 1.1 (Rosenberg 2004) was used to display the data graphically.
We used Arlequin to analyze population structure using Wright’s F-statistics (Wright 1968). This analysis requires the populations and groups of populations be known a priori. Therefore, we used the algorithm to test the hypothesis of populations relationships by clustering population samples into groups based on our mtDNA phylogeny, Structure clusters, or named subspecies and compared the values of the F statistics.
Results
Microsatellite variation was determined for 147 individuals from 18 rivers (Table 2). All four loci were polymorphic: CRAL17 had 19 alleles (136–212 bp), CRAL4 had 17 alleles (161–201 bp), CRAL9 had 13 alleles (119–147 bp), and CRAL13 had 10 alleles (187–205 bp). Our loci showed per population allele numbers ranging from an average of 5.50 alleles (WFGR, CR) to 1.75 alleles (EPR).
Genotype frequencies generally conformed to Hardy–Weinberg equilibrium in all populations across all loci, with one exception at Copper Creek (locus CRAL9). Observed and expected heterozygosities for each locus at all populations are shown in Table 2. Microchecker suggested the presence of null alleles in the Spring River population in one locus (again CRAL9). To test if the null allele or the deviation from Hardy–Weinberg affected our results, we ran the complete dataset for all analyses as well as the dataset excluding the CRAL9 locus in the Spring River and the Copper Creek population. There were only minor differences in the results, so we felt we were able to include this locus in our analyses.
The results of the Structure analysis estimated the most probable value of K to be 11 clusters (Bayesian posterior probability = 0.954). However, when populations are distributed in hierarchical groups, in which migration among populations within a group is greater than migration among groups, Evanno et al. (2005) showed that the most likely value of K may not provide an accurate estimate of the number of higher level groups. To estimate the number of population groups, we employed the method outlined in Evanno et al. (2005) which uses the variance adjusted rate of change in the likelihood of K (∆K) to estimate the number of clusters. Using this approach, the highest ∆K was found at K = 12, with a second peak at K = 8 (Fig. 2). We next compare all three of the possible K values (K = 8, 11, and 12) to the mtDNA results of Sabatino and Routman (2009).

Plot of ΔK for the microsatellite data. Peaks at 8 and 12 suggest that populations are hierarchically grouped into 8 or 12 groups
The mtDNA phylogeny from Sabatino and Routman (2009) resolves eight monophyletic groups. To facilitate a direct comparison of the same number of groups, we first compare the microsatellite Structure results for K = 8 with the mtDNA tree from Sabatino and Routman (2009) (Fig. 3, individual river populations are labeled in Fig. 4, to avoid cluttering Fig. 3). The mtDNA tree showed eight reciprocally monophyletic lineages that corresponded to river drainages or geographic region. These are: (1) Northern Ozarks, (2) Ohio-Susquehanna Rivers, (3) Tennessee River, (4) Copper Creek, (5) North Fork of the White, (6) Spring River, (7) New River and (8) Current-Eleven Point Rivers. Most of these groups are also evident in the microsatellites (Fig. 3). The one exception is the New River sample (two specimens from the New River and six specimens from the West Fork of the Greenbrier River). In the mtDNA analysis, the New River sample is highly divergent, although it does tend to cluster with the Current-Eleven Point Rivers group. In the microsatellite analysis, seven individuals from the New River Sample cluster with the sample from Copper Creek (a Tennessee River tributary), and one individual clusters with the Tennessee River group (other tributaries of the Tennessee River).

A comparison of the clusters estimated with Structure for K = 8 with the phylogeny of mtDNA haplotypes (from Sabatino and Routman 2009). Both analyses suggest that hellbenders populations are divided into 8 groups. Colors/shading in the Structure output represent different genetic clusters. Bars on the chart represent each individual, with the proportion of each color/shading equaling the probability of that individual’s membership in that cluster. Individuals are arranged by population, with populations separated by black lines (population names are shown in Fig. 4, to avoid clutter). The C. a. bishopi subspecies is indicated by black dots on the phylogeny. (Color figure online)
Although when K = 11 (the most likely number of groups according to Structure) individuals have greater ambiguity as to cluster membership, the basic geographic division into eight groups is the same with the exception of only a few individuals within some rivers (Fig. 4). We also compare the Structure results for K = 12 (the most likely K found using the ΔK criterion) with those for K = 11 (Fig. 5). Again, the same basic clustering of populations is found in both analyses, with the exception of the New River drainage populations, which seem to cluster with Ohio River or Beaverdam Creek populations when K = 12 and with the Copper Creek population when K = 11.

A comparison of the clusters estimated with Structure for K = 8 (number of groups found by mtDNA sequencing) versus K = 11 (optimal K from Structure). Interpretation of the charts is as in Fig. 3, with major groups named above the upper chart and individual populations named below the lower chart. Although colors/shading were chosen to enhance readability, color/shading cannot be compared across charts, because clusters are independent in each analysis. The single specimen from the Wabash River is not noted as an individual population, and is the rightmost individual in the Slippery Rock Creek sample in all figures. (Color figure online)

A comparison of the clusters estimated with Structure for K = 12 (highest peak for ΔK method) versus K = 11 (optimal K from Structure). Interpretation of the charts is as in Fig. 5. The two graphs differ in the relationships for the New River drainage populations, indicated by lines connecting the populations
In order to determine which genetic structure is best supported by our microsatellite data, we calculated Wright’s hierarchical F-statistics among-groups (FCT), among-populations-within-groups (FSC), and among populations within the total sample (FST) (Excoffier et al. 1992). We partitioned the data into groups in three ways: 1) using the 8 mitochondrial groups found by Sabatino and Routman (2009), 2) the Structure results from this study that were closest to the mitochondrial data (K = 8 but with the New River populations lumped with Copper Creek), and by named subspecies. For all three group partitions, the FST values were very similar, ranging from 0.42 to 0.44. This is expected because the “group” level is not directly involved in estimating this parameter. The among-group variation increases when data are clustered as either the mtDNA groups (FCT = 0.39) or the Structure groups (FCT = 0.37) as compared to the subspecies grouping (FCT = 0.11). However, the among-populations-within-groups variation (FSC) shows the opposite pattern; the subspecies group shows an increased FSC (0.36) as compared to the mtDNA and Structure groups (FSC = 0.08 and 0.09 respectively). The mtDNA groups or Structure groups have higher between group variance and lower within group variance suggesting that either of these groups better represent genetically differentiated population sets than do the named subspecies.
Discussion
Regardless of the number of clusters considered optimal (K = 8, 11, or 12), Structure analysis of microsatellite variation in the hellbender revealed genetic groups concordant with geographic patterns found in the mtDNA phylogeny (Routman et al. 1994; Sabatino and Routman 2009). The combined mtDNA sequence and microsatellite data suggest that many populations surveyed in this study are genetically distinct. Populations of hellbenders comprise at least eight distinct units: (1) Northern Ozarks, (2) Ohio-Susquehanna Rivers, (3) Tennessee River, (4) Copper Creek, (5) North Fork of the White, (6) Spring River, (7) New River and (8) Current-Eleven Point Rivers. In our microsatellite analysis, there is only one population relationship that is incompatible with the mitochondrial findings: samples from the New River Drainage cluster with populations from rivers that drain into the Tennessee River (when K = 8 or 11) or the Ohio River (when K = 12). In the mitochondrial analysis, the hellbenders from the New River group with the highly divergent Current River/Eleven Point River samples, which comprises the sister clade to the rest of the samples. Because the New River drainage hellbenders belong to a unique cluster when the Structure analysis is rerun with CRAL9 locus is omitted from the samples where null alleles were detected by Microchecker (results not shown), the association between the New River and the Tennessee River/Ohio River populations based on Structure analysis of all four microsatellites might be an artifact. On the other hand, the association could reflect incomplete lineage sorting from the common ancestor of the New River and other populations while the clustering of the New River mtDNA haplotypes with the Current River/Eleven Point haplotypes could reflect a true lack of concordance between the gene tree and the species tree. Regardless of the reason for the discrepancy, the New River has a very different mitochondrial haplotype and so warrants status as separate lineage.
The microsatellites have considerable allele sharing among populations (see Table 3 for an example). This lack of population specific autosomal markers could mean the populations are not as evolutionarily isolated as the mitochondrial phylogeny suggests. However, the complete reciprocal monophyly, high divergence, and geographically structured lineages for the mitochondrial data would mean that if ongoing gene flow were responsible for allele sharing, migration would have to be strongly male biased. According to Nickerson and Mays (1973), Peterson (1987), and Routman (unpublished data), hellbenders show low within-river movement and high philopatry for both genders of adult hellbenders. Gene flow mediated by larvae or juveniles (which may have a higher propensity to be washed downstream) are unlikely to exhibit male bias.
However, sharing of alleles may occur without ongoing gene flow. One explanation for microsatellite allele sharing in the presence of mtDNA isolation among populations is that shared microsatellite alleles may have evolved independently (Culver et al. 2001). Another explanation may be the longer coalescence time for autosomal markers. Coalescence theory (Kingman 1982) predicts that the average time to the common ancestor for a sample of autosomal genes is approximately 4Ne generations, where Ne is the effective population size of the population. Because the mitochondrial genome is effectively haploid and maternally inherited, the Ne is one quarter that of nuclear genes (assuming a 1:1 sex ratio), which means lineages achieve reciprocal monophyly faster for mtDNA than neutral nuclear markers and sharing of alleles may represent the retention of ancestral polymorphisms for nuclear DNA but not for mtDNA (Moore 1995). This does not mean the mtDNA results are misleading, but that recently isolated populations might still be sharing neutral autosomal alleles long after mitochondrial genes have become reciprocally monophyletic.
It is important to note that although there is considerable allele sharing, Structure was able to define clusters (or sets of clusters) that are geographically almost identical to the mitochondrial lineages without a priori information about the geographic location for the samples and with only four loci sampled. These concordant data sets strongly suggest that the eight groups found in this study comprise distinct evolutionary units.
Conservation implications
The congruence of two independent types of markers (mitochondrial and autosomal) suggests that the mitochondrial phylogeny may reflect the underlying phylogeny of the populations of this species. This means that named subspecies are not natural groups and should not be used to make management decisions for hellbenders. Treating each subspecies as a management unit will at best combine very divergent populations and at worst combine populations that are not even each other’s closest relatives.
Instead, most populations in the study are genetically distinct and should be managed as at least eight distinct evolutionary significant units (ESUs). The concordance of hellbender microsatellite groups with mtDNA groups demonstrates that these sets of populations have had time to evolve differences in these (presumably) selectively neutral markers. Because we are using neutral markers, we are measuring changes in allele frequency based on a relatively slow nonadaptive process: genetic drift. It takes less time for selection to create differences among isolated populations compared to genetic drift alone; therefore, these groups could easily have evolved unique local adaptations to their environments. For example, the Spring River population is the only population of hellbenders known to breed in the winter rather than in the fall (Peterson et al. 1989). This means that the eight groups should be managed independently to prevent mixing of populations and the disruption of coadapted gene complexes and possible outbreeding depression.
Conversely, the opposite strategy could be employed in the event of population extinctions. If faced with the scenario where captive breeding and/or reintroduction is required to conserve the entire species or a large part of the species, or to prevent extinction of specific populations, it might be best to intentionally mix individuals from different groups. By doing so, managers will be creating a varied gene pool on which selection may operate. Some individuals might have a reduced fitness as a result of outbreeding depression, but as a whole, the infusion of genetic variation might be what allows the species to adapt. Computer simulations have suggested that this approach is quite successful at reducing inbreeding and maximizing evolutionary potential (Earnhardt 1999). Templeton et al. (1990) used this approach successfully to reintroduce collared lizards to glades in the Missouri Ozarks. In the collared lizard system, potential founders for extinct populations were themselves found in small populations, precluding collection of large numbers. Therefore individuals from genetically distinct lineages were used to form mixed founder groups for recolonization of extinct glades to increase the genetic variation of the founder populations. A similar approach may prove useful and, unfortunately, necessary for the hellbender.
References
Aljanabi SM, Martinez I (1997) Universal and rapid salt-extraction of high quality genomic DNA for PCR-based techniques. Nucl Acids Res 25:4692
Beebee TJC (2005) Conservation genetics of amphibians. Heredity 95:423–427
Briggler JT, Ettling J, Wanner M, Schuette C, Duncan M (2007) Cryptobranchus alleganiensis (hellbender). Chytrid fungus. Herpetol Rev 38(2):174
Chan KMA, Levin SE (2005) Leaky prezygotic isolation and porous genomes: rapid introgression of maternally inherited DNA. Evolution 59(4):720–729
Culver M, Menotti-Raymond MA, O’Brien SJ (2001) Patterns of size homoplasy at 10 microsatellite loci in pumas (Puma concolor). Mol Biol Evol 18(6):1151–1156
DeSalle R, Amato G (2004) The expansion of conservation genetics. Nat Rev Genet 5(9):702–712
Dolman G, Phillips B (2004) Single copy nuclear DNA markers characterized for comparative phylogeography in Australian wet tropics rainforest skinks. Mol Ecol Notes 4(2):185–187
Earnhardt JM (1999) Reintroduction programmes: genetic trade-offs for populations. Anim Conserv 2:279–286
Edwards SV, Beerli P (2000) Perspective: gene divergence, population divergence, and the variance in coalescence time in phylogeographic studies. Evolution 54(6):1839–1854
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131(2):479–491
Gharrett AJ, Smoker WW, Reisenbichler RR, Taylor SG (1999) Outbreeding depression in hybrids between odd-and even-broodyear pink salmon. Aquaculture 173(1–4):117–129
Grobman AB (1943) Notes on salamanders with the description of a new species of Cryptobranchus. Occas Pap Univ Mich Mus Zool 470:1–13
Guimond RW, Hutchison VH (1973) Aquatic respiration: an unusual strategy in the hellbender Cryptobranchus alleganiensis alleganiensis (Daudin). Science 182(4118):1263–1265
Hedrick PW (2001) Conservation genetics: where are we now? Trends Ecol Evol 16(11):629–636
Houlahan JE, Findlay CS, Schmidt BR, Meyer AH, Kuzmin SL (2000) Quantitative evidence for global amphibian population declines. Nature 404(6779):752–755
Hudson RR, Turelli M (2003) Stochasticity overrules the three-times rule: genetic drift, genetic draft, and coalescence times for nuclear loci versus mitochondrial DNA. Evolution 57(1):182–190
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806
Johnson JR, KM Faries, JJ Rabenold, RS Crowhurst, JT Briggler, JB Koppelman and LS Eggert (2009). Polymorphic microsatellite loci for studies of the Ozark hellbender (Cryptobranchus alleganiensis bishopi). Conserv Genet, online first doi:10.1007/s10592-009-9818-z
Kingman JFC (1982) On the genealogy of large populations. J Appl Probab 19A:27–43
Lenormand T (2002) Gene flow and the limits to natural selection. Trends Ecol Evol 17(4):183–189
Mayasich J, Granmaison D, Phillips C (2003) Eastern hellbender status assessment report. Nat Resour Res Inst Tech Rep 9:1–41
Merkle DA, Guttman SI, Nickerson MA (1977) Genetic uniformity throughout the range of the hellbender, Cryptobranchus alleganiensis. Copeia 1977(3):549–553
Moore WS (1995) Inferring phylogenies from mtDNA variation: mitochondrial-gene trees versus nuclear-gene trees. Evolution 49(4):718–726
Moritz C (2002) Strategies to protect biological diversity and the evolutionary processes that sustain it. Syst Biol 51(2):238–254
Nickerson MA, Briggler JT (2007) Harvesting as a factor in population decline of a long-lived salamander; the Ozark hellbender, Cryptobranchus alleganiensis bishopi Grobman. Appl Herpetol 4(3):207–216
Nickerson MA, Mays CE (1973) A study of the Ozark hellbender Cryptobranchus alleganiensis bishopi. Ecology 54(5):1164–1165
Peakall R and P Smouse (2006) GenAlEx 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes, Blackwell Synergy 6: 288–295
Peterson CL (1987) Movement and catchability of the hellbender, Cryptobranchus alleganiensis. J Herpetol 21(3):197–204
Peterson CL, Ingersol CA, Wilkinson RF (1989) Winter breeding of Cryptobranchus alleganiensis bishopi in Arkansas. Copeia 1989:1031–1035
Phillips CA, Humphries WJ (2005) Cryptobranchus alleganiensis (Daudin, 1803). In: Lanoo M (ed) Amphibian declines: The conservation status of United States species, University of California Press, Berkeley, CA, pp 648–651
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155(2):945–959
Raymond M, Rousset F (1995) GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86:248–249
Reed DH, Frankham R (2003) Correlation between fitness and genetic diversity. Conserv Biol 17(1):230–237
Rosenberg NA (2004) Distruct: a program for the graphical display of population structure. Mol Ecol Notes 4(1):137–138
Routman E (1993) Mitochondrial DNA variation in Cryptobranchus alleganiensis, a salamander with extremely low allozyme diversity. Copeia 1993(2):407–416
Routman E, Wu R, Templeton AR (1994) Parsimony, molecular evolution, and biogeography: the case of the North American giant salamander. Evolution 48(6):1799–1809
Sabatino SJ, Routman EJ (2009) Phylogeography and conservation genetics of the hellbender salamander (Cryptobranchus alleganiensis). Conserv Genet 10:1235–1246. doi:10.1007/s10592-008-9655-5
Schneider S, Roessli D, Excoffier L (2000) Arlequin version 2.000. A software for population genetics data analysis. Genetics and biometry laboratory, department of anthropology and ecology. University of Geneva, Geneva, Switzerland
Schwartz MK, Luikart G, Waples RS (2007) Genetic monitoring as a promising tool for conservation and management. Trends Ecol Evol 22(1):25–33
Stuart SN, Chanson JS, Cox NA, Young BE, Rodrigues ASL, Fischman DL, Waller RW (2004) Status and trends of amphibian declines and extinctions worldwide. Science 306(5702):1783–1786
Templeton AR, Shaw K, Routman E, Davis SK (1990) The genetic consequences of habitat fragmentation. Ann Missouri Bot Gard 77:13–27
Van Oosterhout C, Hutchinson WF, Wills DP, Shipley P (2004) Micro-checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4(3):535–538
Wheeler BA, Prosen E, Mathis A, Wilkinson RF (2003) Population declines of a long-lived salamander: a 20+-year study of hellbenders, Cryptobranchus alleganiensis. Biol Conserv 109(1):151–156
Williams RD, Gates JE, Hocutt CH, Taylore GJ (1981) The hellbender: a nongame species in need of management. Wildl Soc Bull 9(2):94–100
Wright S (1968) Evolution and the genetic of populations. University of Chicago Press, Illinois
Acknowledgments
We thank Tammy Lim for help in the field. Jeff Briggler provided specimens from the Eleven Point River. This research was partially supported by a grant from the National Park Service (J8C07070005) to EJR. MT was supported by a National Institutes of Health RISE Fellowship (5R25-GMO59298-10). Many thanks to Victoria Grant for facilitation of funding. Lori Eggert provided microsatellite advice.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tonione, M., Johnson, J.R. & Routman, E.J. Microsatellite analysis supports mitochondrial phylogeography of the hellbender (Cryptobranchus alleganiensis). Genetica 139, 209–219 (2011). https://doi.org/10.1007/s10709-010-9538-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10709-010-9538-9