
Abstract
We report the heritable germ-line transformation of the Queensland fruit fly, Bactrocera tryoni, using a piggyBac vector marked with either the fluorescent protein DsRed or EGFP. A transformation frequency of 5–10% was obtained. Inheritance of the transgenes has remained stable over more than 15 generations despite the presence of endogenous piggyBac sequences in the B. tryoni genome. The sequence of insertion sites shows the usual canonical pattern of piggyBac integraton into TTAA target sites. An investigation of endogenous piggyBac elements in the B. tryoni genome reveals the presence of sequences almost identical to those reported recently for the B. dorsalis complex of fruit flies and two noctuid moths, suggesting a common origin of piggyBac sequences in these species. The availability of transformation protocols for B. tryoni has the potential to deliver improvements in the performance of the Sterile Insect Technique for this pest species.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The development of genetic transformation protocols for non-drosophilid insects has greatly increased the available options for improvement of the Sterile Insect Technique (SIT) to control insect pests. Molecular technologies can be used to introduce genetic markers and develop male-only broods through manipulation of the sex-determination pathway. In Australia, the major pest of fruit production is the Queensland fruit fly, Bactrocera tryoni. Problems with current SIT strains of Queensland fruit fly include the lack of a reliable marker for release strains and lack of a male-only strain. Both of these problems can be addressed by specific genetic manipulation of the species.
Genetic transformation of a range of non-drosophilid insect species has been achieved in recent years with four different transposon vectors (Robinson et al. 2004). The most versatile of these is piggyBac, which has been used in many different species covering more than three orders (Handler 2002). Although piggyBac has been very successful as a transformation vector for a range of insect species, one of the concerns with its use is the potential for instability of the introduced genetic material due to the presence of endogenous piggyBac sequences in the host genome. Indeed, piggyBac-related sequences have been identified in a wide range of organisms (Sarkar et al. 2003), and the Oriental fruit fly, Bactrocera dorsalis, contains multiple piggyBac elements with very strong similarity (95% DNA sequence identity) to the original IFP2 Trichoplusia ni piggyBac element (Handler and McCombs 2000). Despite this, piggyBac was used successfully to transform B. dorsalis without any evidence of instability. Sequences with high similarity to active piggyBac have also been identified in the genomes of a number of related Bactrocera species including the Australian species, B. tryoni, B. neohumeralis and B. jarvisi, although none of the identified sequences encodes a complete transposase (Bonizzoni et al. 2007).
Here we report the genetic transformation of B. tryoni by microinjection of two piggyBac vectors, one marked with the fluorescent protein EGFP, the other with DsRed (Handler and Harrell 2001a, b). We found no alteration to the piggyBac vector or flanking B. tryoni sequences at the insertion sites 15 generations after transgene integration. We have also investigated the presence of endogenous piggyBac elements in the B. tryoni genome using vectorette and inverse PCR protocols that can distinguish different chromosomal insertion sites.
Methods
Microinjections
Embryos were collected from a B. tryoni lemon-eye (le) stock, homozygous for an eye-colour mutation with very light yellow eyes (Zhao et al. 2003). Embryos were dechorionated for 1 min in 1% sodium hypochlorite and rinsed in 0.02% Triton-X100 and milliQ water. Dechorionated embryos were aligned on double-sided tape with the posterior pole towards the edge of the tape and covered by halocarbon oil 700. A mixture of plasmid DNA carrying the piggyBac transposon marked either with DsRed (PUb-DsRed1) or EGFP (PUbnlsEGFP) at 400 ng/μl and the helper plasmid (phsp-pBac) at 200 ng/μl, all in injection buffer (5 mM KCl, 0.1 mM sodium phosphate pH 6.8), were injected into the posterior pole up to 3 h after laying. Following injection, embryos were kept in a humidified atmosphere and, after 24 h, were heat shocked at 37°C for 0, 30 or 60 min. Emerged G0 adult flies were crossed in small groups with le flies. The G1 adults were screened for EGFP or DsRed expression using a Leica MZ FLIII stereomicroscope fitted with GFP2 and DSR filters, and individual fluorescent G1 flies were back-crossed to le flies.
PCR
Flies were tested for the presence of the injected DNA by direct PCR using primers within the EGFP or DsRed coding region:
EGFPfor | 5′ ATGGTGAGCAAGGGCGAGGAG 3′ |
EGFPrev | 5′ CTTGTACAGCTCGTCCATGCC 3′ |
Redfor | 5′ CGTGAAGCTGAAGGTGACCAAG 3′ |
Redrev | 5′ TGCTCCACGATGGTGTAGTC 3′ |
Inverse PCR
Genomic DNA was isolated from adult flies using phenol/chloroform, as described in Shearman and Frommer (1998). Genomic DNA (2 μg) was digested with the restriction enzymes ClaI or TaqI for the 5′ junction, and BstBI or HpaII for the 3′ junction. Each digest was ligated in a volume of 400 μl using T4 DNA ligase (NEB) at 16°C for 16 h. After ethanol precipitation PCR was performed using primers UbIPCR and egfpIPCR (ClaI digests), pBacIPCR51 and pBacIPCR52 in round 1 and pBacIPCR5L and pBacIPCR54 in round 2 (TaqI digests), UbIPCR and DsRedIPCR in round 1 and UbIPCR and pBac3SP1 in round 2 (BstBI digests), pBac3IPCR31 and pBacIPCR32 in round 1 and pBac3IPCR33 and pBacIPCR34 in round 2 (HpaII digests).
UbIPCR | 5′ TGGAATCTCTGTCGCTGCGTTCC 3′ |
egfpIPCR | 5′ GGTCCTGCTGGAGTTCGTGACC 3′ |
DsRedIPCR | 5′ CACCATCGTGGAGCAGTACGAGC 3′ |
pBacIPCR51 | 5′ CACTTACCGCATTGACAAGCACG 3′ |
pBacIPCR52 | 5′ TTTGAACTATAACGACCGCGTGAG 3′ |
pBacIPCR5L | 5′ GATGTCCTAAATGCACAGCGACGG 3′ |
pBacIPCR54 | 5′ CGCATGATTATCTTTTACGTGAC 3′ |
pBac3SP1 | 5′ CAGACCGATAAAACACATGCGTC 3′ |
pBacIPCR31 | 5′ TAAATAAACCTCGATATACAGACCG 3′ |
pBacIPCR32 | 5′ CTTATTAGTCAGTCAGAAACAACTTTG 3′ |
pBacIPCR33 | 5′ ACACATGCGTCAATTTTACGCATG 3′ |
pBacIPCR34 | 5′ GCACATATCAATATTATGCTCTCGAC 3′ |
Vectorette PCR
Genomic DNA was isolated from adult flies using phenol/chloroform, as described in Shearman and Frommer (1998). Vectorette PCR was performed essentially as described in Wang et al. (2006), except that TaqI (NEB) was used to digest genomic DNA, modified primers were used and ligation of the vectorette unit was performed separately to the restriction enzyme digestion. The sequence of the vectorette unit (Link 1 and Link 2) and primers used in vectorette PCR are shown below.
Results and discussion
Transformation experiments
Interplasmid transposition assays have shown that five transposon vectors, including piggyBac, are capable of accurate transposition in B. tryoni eggs (Raphael et al. 2004). Here we report the genetic transformation of B. tryoni using two different piggyBac vectors marked with either the dominant visible markers EGFP or DsRed. Both the PUbnlsEGFP and PUb-DsRed1 constructs were coinjected with the phsp-pBac helper plasmid which drives transposase expression from the D. melanogster hsp70 promoter (Handler and Harrell 1999). Embryos were exposed to a heat shock of 37°C, 24 h post injection. The length of the heat shock had a major effect on the number of adults that emerged from injected eggs (Table 1). A heat shock of 1 h resulted in 2–4% emerged adults compared to 6–8% for a 30-min heat shock and 12% with no heat shock.
Emerged G0 flies were crossed in small groups with le flies and G1 adult progeny screened for EGFP or DsRed fluorescence. The minimum transformation frequency (Table 1) was calculated assuming one fly in each group gave rise to all the G1 fluorescent flies. No transformants were obtained with a 60-min heat shock. A total of 16 fluorescent G1 flies were obtained from DsRed injections, 3 of these with no heat shock of injected embryos (5.3% minimum transformation frequency), 13 with a 30-min heat shock (10.3% minimum transformation frequency). The substantial transformation frequency in the absence of heat shock is indicative of the expected constitutive expression from the heat shock promoter. On the other hand, the relatively lower transformation frequency with no heat shock compared to a 30-min heat shock could suggest lower integration frequency of the transposon due to insufficient expression of the transposase from the Dm-hsp70 promoter, although only one batch of injected eggs received no heat shock treatment. Injection of the EGFP-marked construct resulted in 16 fluorescent G1 flies (5.6% minimum transformation frequency) from a 30-min heat shock.
Individual G1 flies were crossed to le in order to establish lines of transformed flies and to determine the inheritance pattern of the transgene from the proportion of fluorescent G2 offspring (Fig. 1). Six of the 16 DsRed-transformed G1 flies and 9 of the 16 EGFP-transformed G1 flies were fertile. Assuming a single copy of the transposon is inserted into the genome of transformed G1 flies, 50% of the G2 progeny are expected to show the fluorescent phenotype. For the DsRed transformed lines this is the case for 3 of the 6 lines, however lines Red 2, 4 and 6 show a higher than expected frequency of fluorescent flies, close to 70%, suggesting that more than one transgene may have integrated. All but one of the 9 EGFP lines show the expected 50% positive flies, only Green 6 deviates with a frequency of 17.4%. The strength of fluorescence was very low in flies from this line, so the low frequency of positive progeny may reflect difficulties in detection of EGFP fluorescence, potentially due to a genomic position effect (discussed below).

The number of fluorescent (green or red bars) and non-fluorescent (cream bars) G2 progeny from individual G1 flies crossed with B. tryoni le. Asterisks mark a significant deviation from 1:1 ratio of fluorescent: non-fluorescent progeny
Homozygous lines have been created by setting up crosses of single males and females from each line and screening the progeny for fluorescence. Lines with 100% fluorescent offspring were inbred and progeny screened every generation, removing any non-fluorescent flies. After 5 generations four lines (two PUb-DsRed transformed lines and two PUb-nlsEGFP transformed lines) were produced without the appearance of negative flies, and are therefore putatively homozygous. These have been maintained for more than 15 generations, with test screening for 100% fluorescence every second generation, indicating that the marker is stably inherited in the germ-line.
EGFP and DsRed expression patterns
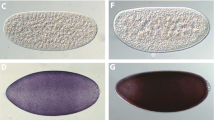
EGFP expression patterns are similar to those reported previously for the same construct in Anastrepha suspensa (Handler and Harrell 2001a), and DsRed expression was similar but with brighter fluorescence compared to EGFP. Most of the lines show a similar pattern of fluorescent protein expression. DsRed is particularly strong and is clearly visible on the dorsal thorax and legs (Fig. 2f). In some flies, red fluorescence is also visible in the abdomen. The eyes do not show any evidence of expression. EGFP is most strongly expressed in the legs (Fig. 2n), occasionally green fluorescence can be seen on the dorsal thorax. The ventral abdomen displays quite strong autofluorescence (Fig. 2j). Similar to DsRed transformants, the eyes do not express EGFP. Both DsRed and EGFP are visible in larvae (Fig. 2h, p), however fluorescence was not detected in 1 day old embryos in any of the lines. One of the DsRed lines (Red 3) and one of the EGFP lines (Green 6) had weak expression and breeding of these lines was discontinued. DsRed but not EGFP fluorescence remained visible 2 months after death.

DsRed and EGFP expression in B. tryoni le adults and larvae. Non transformed adult, dorsal view—a white light and b DSR filter; non transformed larvae—c white light and d DSR filter. Adult transformed with PUb-DsRed1, dorsal view—e white light and f DSR filter; larvae transformed with PUb-DsRed1—g white light and h DSR filter. Non transformed adult, ventral view—i white light and j EGFP2 filter; non transformed larvae—k white light and (l) EGFP2 filter. Adult transformed with PUbnlsEGFP, ventral view—m white light and n EGFP2 filter; larvae transformed with PUbnlsEGFP—o white light and p EGFP2 filter
Some variation in intensity of fluorescence was apparent between different transformed lines, probably due to chromosome position effects, as has been reported previously for similar constructs (Handler and Harrell 2001a). However, no significant differences in expression pattern between lines were observed, in contrast to a similar construct in Ceratitis capitata (Scolari et al. 2008) and EGFP expression driven by the tetracycline operator in B. oleae (Koukidou et al. 2006).
Insertion sites of transgenic DNA
The presence of transgenic DNA in each of the lines has been confirmed by PCR using primers within the EGFP and DsRed coding regions of the plasmids. Inverse PCR was used to characterise the insertion sites in four of the lines (Online Resource Fig. S1). All show the TTAA motif at each junction site characteristic of piggyBac insertion. The insertion sites were differentiated from endogenous piggyBac elements by amplification using a primer designed to the genomic DNA at the left and right insertion sites (from sequence determined by inverse PCR, see Online Resource Fig. S1), together with a primer from within the transgene, either the EGFP or DsRed sequence. The integrity of the transgenic DNA and the insertion sites were confirmed by PCR using these primers on genomic DNA isolated from flies 15 generations from the original G1 fly. In addition, it was confirmed that the two primers at the left and right insertion sites amplified a genomic DNA band of the correct size in the le background strain.
Endogenous piggyBac sequences in the B. tryoni lemon-eye strain
Inverse PCR, direct PCR and a vectorette PCR method (Wang et al. 2006), designed to amplify the left and right piggyBac arms and flanking genomic DNA, were used to characterise some endogenous piggyBac elements in the le strain of B. tryoni. We found endogenous piggyBac sequences that are very closely related to the original IFP2 piggyBac transposon, and to sequences recently isolated from the B. dorsalis complex of Tephritid flies and noctuid moths (Handler et al. 2008; Zimowska and Handler 2006). The majority of the base changes in the le sequences compared to T. ni piggyBac are also present in the B. dorsalis complex consensus sequence (Online Resource Fig. S2a and Fig. S3), including 10 base changes leading to a change in the conceptual amino acid translation of the transposase, suggesting that the B tryoni and B. dorsalis sequences share a common origin. One of the regions isolated encodes the first 62 amino acids, and a second region encodes 149 amino acids, of a transposase each with >90% amino acid identity to the equivalent regions of the T. ni transposase. It is unknown if a complete open reading frame is present. Two endogenous le sequences have deletions of more than 90 nucleotides in the region upstream from the transposase coding region. The deletions are likely to affect the promoter and therefore gene expression. Other, direct PCR products confirm the presence of at least four deleted piggyBac elements in the le genome.
A small fragment of DNA from the right arm of piggyBac and the adjacent genomic DNA was amplified from le genomic DNA (Online Resource Fig. S2b). The 52 nucleotides of piggyBac sequence show complete identity with the T. ni sequence including the inverted terminal repeat and 8 nucleotides of the sub-terminal repeat. The adjacent genomic DNA contains the TTAA sequence, the expected target site of the piggyBac transposon.
These results add to a previous investigation in which PCR amplification and Southern blotting revealed the presence of four piggyBac sequences in the genome of B. tryoni (Bonizzoni et al. 2007), in agreement with our PCR results. The sequences reported by Bonizzoni et al. are from within the transposase coding region and do not overlap with those reported here.
This is the first report of the germ-line transformation of the Australian tephritid fruit fly, B. tryoni. using the piggyBac vector. B. tryoni has been transformed previously with a hobo vector (Atkinson et al. 1996), however molecular analysis revealed imperfect transposition and instability, with loss of the hobo element in subsequent generations (S. Whyard, reported in Raphael et al. 2004). In the case of the piggyBac vector reported here, the transgenic DNA has been stably inherited for more than 15 generations with all insertion sites examined showing the presence of the left and right piggyBac inverted repeat and the TTAA motif at each junction site, characteristic of piggyBac insertion. The results indicate that piggyBac represents a feasible transformation vector for long-term genetic transformation, despite the presence of endogenous piggyBac derivatives in the genome. We note that endogenous piggyBac sequences are degraded and present at low frequency (less than 10 copies per genome), by contrast, for example, with the high copy number of mariner-related sequences in the B. tryoni genome (Green and Frommer 2001). Nevertheless, the possibility of interaction between endogenous piggyBac sequences and an introduced vector via the inverted terminal repeats (ITR) remains. Therefore future work will include the post-integration stabilisation of the piggyBac vector by removal of one of the ITR sequences (Handler et al. 2004).
Currently, SIT is the only non-pesticide option for complete eradication of pest populations on an area-wide basis. Together with increases in the fitness of the Qfly SIT strains by improved breeding methods, transformation technology has the potential to deliver improvements in the performance of SIT as a sustainable pest control measure for Queensland fruit fly. We have now generated transformed lines with single well-characterised insertion sites of piggyBac using attP (Groth et al. 2004; Scolari et al. 2008) and this will permit qualitative and quantitative analysis of a range of genetic constructs in the B. tryoni genome. These will include endogenous markers to replace the current practice of dusting the mass-reared flies externally with fluorescent dyes, which are a health hazard and are prone to error in field monitoring, and constructs that allow manipulation of the sex-determination pathway to produce male-only strains, as in the use of RNA interference constructs to inhibit expression of transformer in fertilised eggs (Pane et al. 2002). Genetic transformation will also be useful for functional genetic studies such as the role of circadian genes in species differentiation and pest status (An et al. 2002, 2004).
References
An X, Wilkes K, Bastian Y, Morrow JL, Frommer M, Raphael KA (2002) The period gene in two species of tephritid fruit fly differentiated by mating behaviour. Insect Mol Biol 11:419–430
An X, Tebo M, Sunmi S, Frommer M, Raphael KA (2004) The cryptochrome (cry) gene and a mating isolation mechanism in tephritid fruit flies. Genetics 168:2025–2036
Atkinson PW, Warren WD, Whyard S, Saville KJ, Mende HA, Pinkerton AC, Coates CJ, O’Brochta DA (1996) Mobility of hobo transposable elements in non-drosophilid insects. In: International Atomic Energy Agency Technical Documents Series: Genetic Engineering Technology for the Improvement of the Sterile Insect Technique
Bonizzoni M, Gomulski LM, Malacrida AR, Capy P, Gasperi G (2007) Highly similar piggyBac transposase-like sequences from various Bactrocera (Diptera, Tephritidae) species. Insect Mol Biol 16:645–650
Green CL, Frommer M (2001) The genome of the Queensland fruit fly Bactrocera tryoni contains multiple representatives of the mariner family of transposable elements. Insect Mol Biol 10:371–386
Groth AC, Fish M, Nusse R, Calos MP (2004) Construction of transgenic Drosophila by using the site-specific integrase from phage ΦC31. Genetics 166:1775–1782
Handler AM (2002) Use of the piggyBac transposon for germ-line transformation of insects. Insect Biochem Mol Biol 32:1211–1220
Handler AM, Harrell RA (1999) Germline transformation of Drosophila melanogaster with the piggyBac transposon vector. Insect Mol Biol 8:449–458
Handler AM, Harrell RA (2001a) Transformation of the Caribbean fruit fly, Anastrepha suspensa, with a piggyBac vector marked with polyubiquitin-regulated GFP. Insect Biochem Mol Biol 31:199–205
Handler AM, Harrell RA (2001b) Polyubiquitin-regulated DsRed marker for transgenic insects. Biotechniques 31:820–828
Handler AM, McCombs SD (2000) The piggyBac transposon mediates germ-line transformation in the Oriental fruit fly and closely related elements exist in its genome. Insect Mol Biol 9:605–612
Handler AM, Zimowska GJ, Horn C (2004) Post-integration stabilisation of a transposon vector by terminal sequence deletion in Drosophila melanogaster. Nat Biotechnol 22:1150–1154
Handler AM, Zimowska GJ, Armstrong KF (2008) Highly similar piggyBac elements in Bactrocera that share a common lineage with elements in noctuid moths. Insect Mol Biol 17:387–393
Koukidou M, Kinakis A, Reboulakis C, Zagorailou L, Tavernarkis N, Livadaras I, Economopoulos A, Savakis C (2006) Germ Line transformation of the olive fly Bactrocera aleae using a versatile transgenesis marker. Insect Mol Biol 15:95–103
Pane A, Salvemini M, Bovi PD, Polito C, Saccone G (2002) The transformer gene in Ceratitis capitata provides a genetic basis for selecting and remembering the sexual fate. Development 129:3715–3725
Raphael KA, Whyard S, Shearman D, An X, Frommer M (2004) Bactrocera tryoni and closely related pest tephritids–molecular analysis and prospects for transgenic control strategies. Insect Biochem Mol Biol 34:167–176
Robinson AS, Franz G, Atkinson PW (2004) Insect transgenesis and its potential role in agriculture and human health. Insect Biochem Molec Biol 34:113–120
Sarkar A, Sim C, Hong Y-S, Hogan JR, Fraser MJ, Robertson HM et al (2003) Molecular evolutionary analysis of the widespread piggyBac transposon family and related domesticated sequences. Mol Genet Genomics 270:173–180
Scolari F, Schetelig MF, Bertin S, Malacrida AR, Gasperi G, Wimmer EA (2008) Fluorescent sperm marking to improve the fight against the pest insect Ceratitus capitata (Wiedemann; Diptera: Tephritidae). New Biotechnol 25:76–84
Shearman DCA, Frommer M (1998) The Bactrocera tryoni homologue of the Drosophila melanogaster sex-determination gene doublesex. Insect Mol Biol 7:355–366
Wang J, Miller TA, Park Y (2006) Development of multiple dominant markers by using Vectorette PCR-based nonradioactive transposable element display. Mol Ecol Notes 6:642–645
Zhao JT, Bennett CL, Stuart G, Frommer M, Raphael KA (2003) The scarlet eye-colour gene of the tephritid fruit fly Bactrocera tryoni and nature of two eye-colour mutations. Insect Mol Biol 12:263–269
Zimowska GJ, Handler AM (2006) Highly conserved piggyBac elements in noctuid species of Lepidoptera. Insect Biochem Mol Biol 36:421–428
Acknowledgments
We thank Stuart Gilchrist and Alfie Meats for assistance and advice on fly rearing, and Prof. Steve Simpson for use of the Leica MZ FLIII stereomicroscope. This research was supported by an Australian Research Council Linkage grant LP0667914, with support from the Victorian Department of Primary Industries, the Department of Primary Industry and Resources of South Australia, Griffith City Council, Riverina Citrus and Cobar Shire Council.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Fig. S1
Genomic sequence at the insertion sites of transgenic lines Green 1, Green 8, Red 1 and Red 5. TTAA in bold, genomic sequence in capital letters, piggyBac sequence in lower case. Primers designed to genomic DNA and used to amplify across the junction are underlined. (DOC 21 kb)
Fig. S2
Alignment of B. tryoni piggyBac sequences amplified from le genomic DNA with T. ni piggyBac. a. The first 514 nt of the IFP2 T. ni piggyBac transposon and the first 62 amino acids of the transposase aligned with the consensus of five piggyBac sequences amplified from four B. dorsalis complex species (Genbank accession numbers: EU139843, EU139844, EU139845, EU139846, EU139847) and two sequences amplified from le genomic DNA. Nucleotide mismatches are shown in orange. Changes to the conceptual amino acid translation are shown in red beneath the alignment. A single base pair difference at position 42 from a T in IFP2 to a C in le11 (also a C in the B. dorsalis complex consensus sequence) introduces a TaqI site not present in the original T. ni sequence. TaqI was the Restriction Enzyme used in the vectorette method, consequently no sequence upstream of the TaqI site, including the adjacent genomic DNA, was obtained. b. The 3’ arm of the piggyBac transposon aligned with le sequence. Yellow highlight is the inverted terminal repeat. Green highlight is the sub terminal repeat. The TTAA sequence is in bold. (DOC 36 kb)
Fig. S3
Alignment of a 449 bp piggyBac sequence amplified from B. tryoni le genomic DNA (two independent and identical PCR products) with the T. ni transposon and the consensus of four B. dorsalis sequences (EU139547, EU139549, EU139554, EU139559). The alignment corresponds to nt 1617-2065 of the IFP2 transposon sequence. Nucleotide mismatches are shown in orange, changes to the conceptual amino acid translation are shown in red beneath the alignment. (DOC 54 kb)
Rights and permissions
About this article
Cite this article
Raphael, K.A., Shearman, D.C.A., Streamer, K. et al. Germ-line transformation of the Queensland fruit fly, Bactrocera tryoni, using a piggyBac vector in the presence of endogenous piggyBac elements. Genetica 139, 91–97 (2011). https://doi.org/10.1007/s10709-010-9500-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10709-010-9500-x