
Abstract
The small heat shock proteins (sHSPs) are a ubiquitous family of molecular chaperones. We have identified 18 sHSPs in the Caenorhabditis elegans genome and 20 sHSPs in the Caenorhabditis briggsae genome. Analysis of phylogenetic relationships and evolutionary dynamics of the sHSPs in these two genomes reveals a very complex pattern of evolution. The sHSPs in C. elegans and C. briggsae do not display clear orthologous relationships with other invertebrate sHSPs. But many sHSPs in C. elegans have orthologs in C. briggsae. One group of sHSPs, the HSP16s, has a very unusual evolutionary history. Although there are a number of HSP16s in both the C. elegans and C. briggsae genomes, none of the HSP16s display orthologous relationships across these two species. The HSP16s have an unusual gene pair structure and a complex evolutionary history shaped by gene duplication, gene conversion, and purifying selection. We found no evidence of recent positive selection acting on any of the sHSPs in C. elegans or in C. briggsae. There is also no evidence of functional divergence within the pairs of orthologous C. elegans and C. briggsae sHSPs. However, the evolutionary patterns do suggest that functional divergence has occurred between the sHSPs in C. elegans and C. briggsae and the sHSPs in more distantly related invertebrates.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The small heat shock proteins (sHSPs) are a ubiquitous family of proteins that is both diverse and ancient (Caspers et al. 1995; de Jong et al. 1998; Franck et al. 2004; Fu et al. 2006). sHSP homologs are present in eukaryotes, bacteria, and archaea; thus, this family originated before the divergence of the three domains of life. In general, the sHSP monomers are composed of a variable N-terminal region and a more conserved C-terminal region. The C-terminal region is frequently referred to as the α-crystallin domain. This name comes from one of best-studied members of the large sHSP family, the α-crystallin lens proteins of the vertebrate eye. The α-crystallins, which are found only in the lens, evolved via gene duplication from a heat-inducible sHSP (Piatigorsky and Wistow 1989, 1991). Gene duplication has thus played an important role in sHSP evolution. In addition, studies of the sHSPs have also been extremely influential in the development of theories of gene family evolution. The evolution of the α-crystallins was one of the first documented cases of gene sharing (Piatigorsky and Wistow 1989, 1991). Gene sharing is when new protein function evolves before gene duplication, and for a period of time a single protein has two distinct functions. In this case, a sHSP evolved a new function as an eye lens protein prior to gene duplication (Piatigorsky and Wistow 1989, 1991). This finding was instrumental to the development of more recent theories of gene family evolution including evolution by subfunctionalization (Force et al. 1999; Lynch and Force 2000).
The diversity of the sHSPs is quite high, with only a handful of amino acid residues conserved across all known sHSPs (de Jong et al. 1998; Waters and Vierling 1999; Franck et al. 2004). This is quite surprising when compared to the high conservation seen among the other heat shock proteins including the HSP70s (Boorstein et al. 1994). Yet despite this high level of amino acid sequence diversity there is considerable structural conservation among the sHSPs (Kim et al. 1998; van Montfort et al. 2001). Comparisons of the two known sHSP crystal structures, one from the archaea Methanococcus jannaschii (HSP16.5) and the other from the plant Triticum aestivum (HSP16.9), reveals a conserved structural organization. Both proteins form large oligomers (HSP16.5 is a 24-mer and HSP16.9 a dodecamer) from a shared β-sheet sandwich building block. Secondary structures found in the C-terminal regions or α-crystallin domains of both proteins are also highly conserved (van Montfort et al. 2002). The N-terminal regions are not completely resolved in the crystal structures, and this region is also highly diverse across the sHSP family. However, it has been established that the N-terminal domain is needed for oligomer formation (van Montfort et al. 2002).
Numerous studies have established that the sHSPs are molecular chaperones (van Montfort et al. 2002; Haslbeck et al. 2005; Sun and MacRae 2005). They are able to bind to denatured proteins and thus prevent irreversible aggregation (van Montfort et al. 2002; Haslbeck et al. 2005; Sun and MacRae 2005). To date there is no evidence of high levels of substrate specificity among the sHSPs, and they are able to bind to a large number of different proteins at the same time. Developing models of the chaperone network suggests that the sHSPs work with a variety of other chaperones and that denatured proteins can be transferred from one chaperone to another during refolding (Lee and Vierling 2000; Haslbeck et al. 2005). There is however some evidence that some of the most divergent members of sHSP family may not share the conserved oligomeric structure or chaperone function (van Montfort et al. 2002; Haslbeck et al. 2005). The best-studied and most interesting of these divergent proteins include the 12 kDa sHSPs from Caenorhabditis elegans (Leroux et al. 1997a, b).
The presence of diverse sHSPs in C. elegans raises some very interesting evolutionary questions. For example, when and how did they evolve and do they have homologs in other species? The release of the complete genomes of both Caenorhabditis elegans and its close relative Caenorhabditis briggsae now enables us to address a number of questions concerning the evolution of the sHSPs. This species pair has been used successfully to study the evolution of other gene families, including the HSP70s (Nikolaidis and Nei 2004). These species are quite interesting. It is estimated that they diverged between 80 and 100 million years ago (Stein et al. 2003). Yet they are indistinguishable by eye, have the same genome size and chromosome number, share considerable synteny across the chromosomes, and have the same ecological niche.
In our study we have found very complex patterns of sHSP evolution. The sHSPs in C. elegans and C. briggsae do not display clear orthologous relationships with other invertebrate sHSPs. However, many but not all sHSPs in C. elegans have orthologs in C. briggsae. One large group of sHSPs that does not have clear C. briggsae: C. elegans orthologs are the HSP16s. The HSP16s have a complex evolutionary history shaped by gene duplication, gene conversion, and purifying selection.
Material and methods
Identification of sHSPs in the Caenorhabditis elegans and Caenorhabditis briggsae genomes
C. elegans and C. briggsae genome databases at Wormbase.org (v. WS176) were searched using the blastp and blastn (Altschul et al. 1997) programs available at these genome websites. Known C. elegans sHSPs were used as query sequences. We evaluated all sequences that had an e score of 1.0 × 10−5 or lower. These newly identified sequences were then used as queries in additional searches until no new sHSPs were identified. Chromosome location was also obtained for each sHSP from the genome databases. We eliminated isoforms by visually inspecting alignments and then comparing genomic location data. The final list of C. elegans and C. briggsae sequences are given in Table 1A and B.
Sequences from other genomes were likewise retrieved from the NCBI website (http://www.ncbi.nih.gov/Genomes/). The complete genomes of Apis mellifera, Drosophila melanogaster, Anopheles gambiae, and Saccharomyces cerevisiae were searched using blastp v.2.2.10, again using C. elegans sHSPs as query sequences. Sequences were again judged by an e score cutoff 1.0 × 10−5. Imported sequences were then verified through visual inspection of the conserved domain. Sequence identification numbers are given in Table 2.
Initial amino acid alignments were generated with ClustalW (Higgins et al. 1996) using default parameters. The alignments were then manually optimized if necessary in Bioedit v7.0.5 (Hall 1999). Next, those amino acid alignments were used as templates to align the DNA sequences. The DNA alignments were generated using align2aa (http://www.sunflower.bio.Indiana.edu/∼wfischer/Perl_Scripts) and are available from the authors. Secondary structures (found in Fig. 1) were predicted for the C. elegans and C. briggsae proteins using the program PedictProtein (Rost et al. 2004).

Alignment of the conserved carboxyl-terminal region of the C. elegans and C. briggsae sHSPs. The β-sheets are based on secondary structure predictions. The numbering system corresponds to the known secondary structure of HSP16.9 from T. aestivum (van Montfort et al. 2002)
Phylogenetic and evolutionary analysis
Phylogenetic tree construction began with evolutionary model testing. Model testing on DNA alignments was done using Mrmodeltest version 2.2 (Posada and Buckley 2004). Mrmodeltest assigned models of evolution for each nucleotide position in a codon. Model testing for the amino acid alignments was done within MrBayes 3.1 (Ronquist and Huelsenbeck 2003) using the mixed model option that samples from the distributions of 10 different amino acid models of evolution and then provides information on the best model for a given alignment. In our analysis the WAG model was the best fit for the amino acid alignments, and a GTR + I + g model was the best fit for each of the codon positions the DNA data. Phylogenetic trees were then generated using MrBayes version 3.1.
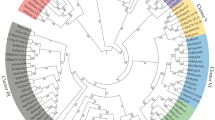
Phylogenetic trees were constructed from an amino acid alignment (available from the authors) of the C. elegans, C. briggsae and other invertebrate sHSP sequences (presented in Fig. 2). This analysis was conducted to evaluate how the sHSP genes from the C. elegans and C. briggsae genomes are related to sHSPs from other invertebrates. Final trees were constructed from 3 million generations of four Monte Carlo Markov Chains using the WAG model of protein evolution. As per the manual for MrBayes3.1 (Ronquist and Huelsenbeck 2003) a standard deviation below 0.001 was considered evidence for statistical convergence of the different chains. The first 500,000 generations (before statistical convergence was reached) were discarded as burnin. The consensus tree was rooted using HSP26 from S. cerevisiae.

Phylogenetic tree of the C. elegans and C. briggsae sHSPs with sHSP homologs from other invertebrates. The tree is rooted with HSP26 from S. cerevisiae. The tree was constructed with Bayesian methods implemented in MrBayes. The support vales or posterior probabilities are placed close to each branch. The highest possible support is 1.00. The two major branches that include the C. elegans and C. briggsae sHSPs are labeled 1 and 2. Each of these two major lineages contains both C. elegans and C. briggsae sHSPs and sHSPs from other species. Accession number for the sequences are available in Tables 1A, B and 2
The phylogenetic tree of just the C. elegans and C. briggsae sHSPs (presented in Fig. 3) was constructed from a partitioned dataset of both the DNA and protein alignments using a GTR + I + g model for each codon position for the DNA data, and the WAG model of protein evolution for the amino acid data. A consensus tree was generated from 2.5 million generations of four Monte Carlo Markov Chains. Again, the first 500,000 generations (before statistical convergence was reached) were discarded as burnin. The consensus tree was rooted with HSP20B.

Phylogenetic analysis of C. elegans and C. briggsae sHSPs. This tree is based on a Bayesian analysis of a combined DNA and amino acid alignment. The support values (posterior probabilities) are above each branch. The highest possible support is 1.00. The two major HSP16 lineages II and I are noted in shaded boxes. The branches outside these boxes include the orthologous gene pairs that contain one gene in C. elegans and in C. briggsae
Tests for gene conversion were performed using Geneconv (Sawyer 1999) (http://www.math.wustl.edu/∼sawyer/geneconv/). Analysis was conducted in two ways. First, only silent polymorphisms were examined when looking for gene conversion events. In the second analysis, silent and non-silent polymorphisms were used. Parameters used in both sets of analyses included a common starting point (w123), listing pairwise hits (lp), and a G-scale value of 2. The G-scale allows for the occurrence of substitutions after the recombination event. Statistical significance was estimated by simulated global p-values (derived from the 10,000 permutations) of p < 0.05 in the case of coding sequences, and p < 0.01 in the case of non-coding alignments (Drouin 2002; Mondragon-Palomino and Gaut 2005).
A key measure of amino acid evolution is estimated by the ratio (ω) of nonsynonymous (Ka) to synonymous (Ks) substitutions at the nucleotide level between two homologous proteins. Nonsynonymous substitutions are those that alter the amino acid sequence. Synonymous substitutions do not alter the amino acid sequence. The ratio (ω) is calculated by dividing nonsynonymous by synonymous changes (Ka/Ks). A ω < 1 is indicative of purifying selection, or strong selective to remove amino acid substitutions. A ω = 1 signifies neutral evolution. A ω > 1 indicates positive selection because nonsynonymous substitutions are occurring at a higher rate compared to synonymous substitutions. This suggests the possibility of a new protein function.
Synonymous (Ks) and nonsynonymous (Ka) rates were estimated with the Kumar method available in the MEGA3.1 (Kumar et al. 2004). Further statistical tests for positive selection included analysis with PAML (Yang 1997). Statistical analyses were done using maximum likelihood comparisons of different models of evolution within PAML (Yang 1997). Site-Specific and Branch-Site based likelihood analyses were used. We found no evidence of positive selection among any of the sHSPs in C. elegans or C. briggsae.
Results
The sHSPs genes of C. elegans and C. briggsae
In total, we identified 18 sHSPs in C. elegans (Table 1A, and 20 sHSPs in C. briggsae (Table 1B). Examination of the sHSP amino acid alignments confirmed the presence of conserved consensus motifs associated with secondary structural features found in all sHSPs (Fig. 1). These motifs included F-x-polar-aromatic-x-L-P in β5-6, and polar-G-V-L-polar-aliphatic-polar-aliphatic-P-basic located in β9 (Fig. 1) (Caspers et al. 1995; de Jong et al. 1998). This analysis confirmed that the proteins under study are in fact sHSPs.
The sHSP genes were found on five of the six C. elegans and C. briggsae chromosomes. All nine HSP16 genes were located on the fifth chromosome. Three other sHSP genes were located on the third chromosome (HSP20A, HSP20B, and HSP12.2). An additional three were on the fourth (HSP12.3, HSP12.6, and HSP21). Finally, two were on the X-chromosome (HSP43, HSP25), and one was on the first chromosome (HSP12.1). There were no sHSP genes on chromosome 2.
The C. elegans and C. briggsae sHSPs are not orthologs of known invertebrate sHSPs
Phylogenetic analysis of the C. elegans and C. briggsae sHSPs with homologs from other complete invertebrate genomes revealed that there were no clear orthologous groups (Fig. 2). This pattern suggested that there has been frequent gene duplication and loss within the sHSP family. This is in contrast to a HSP70 tree, which included many of the same species, that did have orthologous groups (Nikolaidis and Nei 2004). It was also clear from our analysis of the sHSPs that there are far more sHSPs in both C. elegans and C. briggsae compared to the numbers found in bee, drosophila, or mosquito. This was despite the fact that the C. elegans and C. briggsae genomes were not larger than these other genomes. From the tree presented in Fig. 2 we noted that there are two major lineages of C. elegans and C. briggsae sHSPs. One lineage (labeled 2 in Fig. 2) included the 43 kDa and the 12 kDa sHSPs; these proteins may be related to a group of sHSPs from D. melanogaster. However, the branch that unites all these proteins was not well supported. The other, much larger lineage (labeled 1) included sHSPs from three other genomes (A. mellifera, D. melanogaster, and A. gambiae) as well a number of different C. elegans and C. briggsae sHSPs. Most notably this lineage included the C. elegans and C. briggsae 16 kDa sHSPs. There were eighteen HSP16 genes in total: eight C. elegans genes and ten C. briggsae genes.
To examine more closely the relationships of the C. elegans and C. briggsae proteins to each other, we constructed a tree that included only the sHSPs from these genomes and was based on both DNA and amino acid sequence data (Fig. 3). One of the most striking features of this tree was the well-supported one-to-one orthologous relationship for most of the sHSPs. Most notably this was seen with the HSP12s, HSP20s, HSP25s, and HSP43s. These patterns indicated that the duplications that generated these sHSPs occurred before the divergence of C. elegans and C. briggsae but after the divergence of the common ancestor with insects. The other striking feature of this tree was that while the HSP16 lineage contains both C. elegans and C. briggsae homologs there are no orthologous groupings within this lineage. Instead, there were two well-supported lineages that contain both C. elegans and C. briggsae sHSPs. We have labeled these lineages the type I and type II HSP16 lineages.
Strong purifying selection is the dominant evolutionary force on the orthologous sHSPs
When genes in a single genome evolve under purifying selection, or selection to maintain function, Ka values are expected to be much lower than Ks values. This pattern was found for the sHSPs found in orthologous pairs (Table 3). For example, in a comparison to the HSP12.6 orthologous genes (that is, HSP12.6 in C. elegans compared to HSP12.6 in C. briggsae), the Ka is 0.08 and the Ks is 0.6, and for the HSP12.3 orthologous genes the Ka is 0.08 and Ks is 0.50. The relationship of Ka to Ks is frequently summarized with the ω-value (ω = Ka/Ks), and the ω-values were almost all very low. Therefore, it can be concluded that these orthologous genes are evolving under purifying selection and that there has been little functional change in these genes since the divergence of C. elegans and C. briggsae.
When genes evolve independently, high Ka values and high Ks values are expected. This pattern was found in the analysis of paralogous groups within these lineages. For instance, when comparing the C. elegans genes HSP17 and C. elegans HSP25, a Ka value of 0.93 and a Ks value of 1.4 were found. This pattern was found for most of the paralogous groups suggesting independent evolution and divergent function. The independent evolution of these genes was confirmed by the analysis with the program Geneconv, which looked for but found no evidence of gene conversion among the genes that are members of C. elegans and C. briggsae orthologous pairs. A very different pattern of sequence evolution was found among the HSP16 genes in C. elegans and C. briggsae.
The HSP16s of both C. elegans and C. briggsae are part of gene pairs
Most of the sHSPs were dispersed across the genomes (Tables 1A and 2). However, the HSP16 genes in both the C. elegans and C. briggsae genomes were found linked in a duplicate gene structure (Fig. 4). Previous studies of the HSP16 genes in C. elegans indicated that all the HSP16 genes in C. elegans are functional (Hong et al. 2004). Detailed biochemical data for the C. briggsae HP16 genes are currently not available but from our sequence analysis it appears that these genes are functional as well. In both species the HSP16 genes were all on chromosome 5. There were three groups of gene pairs (each with the genes in opposite orientations) and two single genes (16A and 16B). All ten C. briggsae HSP16 genes were in gene pairs with opposite orientations (Fig. 4). It is interesting that the gene pairs were not closely related to each other. In fact, each pair contained one gene from the type I HSP16 lineage and one from the type II HSP16 lineage. Each C. briggsae gene pair has at least one snRNA U1 gene. In C. elegans two of the three HSP16 gene pairs have an snRNA U1 gene; an additional snRNA U1 gene is placed in between 16B and 16A.

Genomic organization of the HSP16 genes in C. elegans and C. briggsae. (a) C. elegans HSP16 genes on chromosome 5. (b) C. briggsae HSP16 genes on chromosome 5. The genes that are members of the HSP16 I lineages are in blue, the members of lineage II is in gray. The direction of gene transcription is indicated by the arrows (based on data from Hong et al. 2004). The snRNA U1 genes are noted by the black arrowheads and the placement of snRNAs based on Latchman (1988)
Each HSP16 gene pair contained a gene from the type I HSP16 lineage and one from the type II HSP16 lineage. (Figs. 3 and 4). The I and II HSP16s were quite distinct from each other. The Ka values between genes within a single gene pair (that is, the gene from lineage I vs. the gene from lineage II) were all at least 0.45, and the Ks values are near or over 1.0 (Table 4). Analysis of the Ka and Ks values within HSP16 lineages revealed some very complex evolutionary patterns (Table 5). In many cases the genes within each lineage had relatively high Ka and Ks values. For example, in C. briggsae 16A and 16D were both within the II lineage and have a Ka of 0.30 and a Ks of 0.98. This would indicate that the gene duplication that generated these two genes occurred some time ago and that these genes have been evolving independently. However, there are other cases where there was little to no sequence divergence from one gene pair to another. For example, the second and third gene pairs in C. elegans were identical to each other, even at synonymous sites. In addition, there are very few nonsynonymous substitutions (0.03) between gene pairs 1 and 2, but the Ks value of 0.27 suggests that some time has elapsed since these genes duplicated or recombined. Among the C. briggsae HSP16 genes the Ka and Ks values indicated that three of the five gene pairs had been evolving independently for some time.
To gain a better understanding of the role of gene conversion on the evolution of the HSP16 genes, we used the program Geneconv to identify possible gene conversion events within each genome. We found a small gene conversion tract between gene pairs 1 and 3 in C. elegans (Table 6). But the relatively low Ka between these pairs with a much higher Ks and no long gene conversion tracts, indicated that it is selection and not gene conversion maintaining sequence similarity. We did find a very significant and long gene conversion tract (of 1,113 bp) between gene pairs 3 and 5 in C. briggsae (Table 6). This gene conversion tract ends at a single base pair frame shift that alters the last seven codons of the HSP16F gene.
Discussion
The sHSPs are a ubiquitous family of proteins that form large oligomers and function as molecular chaperones. They are crucial in the heat shock response but are also expressed under a number of other stress conditions and during development (van Montfort et al. 2002; Sun and MacRae 2005; Nakamoto and Vigh 2007). They have had a long and complex evolutionary history (Plesofsky-Vig et al. 1992; Caspers et al. 1995; Waters 1995; de Jong et al. 1998; Franck et al. 2004). The goals of this analysis were to identify the sHSPs in the complete genomes of C. elegans and C. briggsae and to examine their evolutionary dynamics in light of current theories of gene family evolution.
Studies of gene family evolution trace back to the work of Ohno (1970). But the more recent availability of complete genomes has greatly enhanced our ability to study gene family evolution. A long-standing discussion within studies of gene family evolution has been the relative importance of gene conversion versus frequent gene duplication and loss (referred to as birth-and-death processes) in shaping gene families (Nei and Rooney 2005). Another current controversy relates to the relative importance of neofunctionalization and subfunctionalization in maintaining gene duplicates and in the evolution of new protein functions (Lynch and Force 2000; Lynch and Katju 2004). The detection of high levels of nonsynonymous substitutions is usually accepted as evidence of neofunctionalization and changes in gene expression as evidence of subfunctionalization. In this analysis we have found a very complex pattern of gene family evolution that suggests that all these processes have influenced sHSP evolution.
Analysis of other gene families in C. elegans and C. briggsae has found two very different patterns of evolution: divergent evolution and frequent birth-and-death (Nikolaidis and Nei 2004; Sheps et al. 2004; Zhao et al. 2007). The first is seen in the evolution of the MT and ER subfamilies of the HSP70s (Nikolaidis and Nei 2004). The endoplasmic reticulum or ER HSP70s from even very distantly related species (Saccharomyces, Drosophila, Caenorhabditis) form a monophyletic group and the branching patterns reflect species relationships, i.e., they are orthologs. This pattern is also seen among the mitochondrial or MT HSP70s. In our analysis of the sHSPs we found that the sHSPs in C. elegans and C. briggsae are not orthologs of the sHSPs from more distantly related species such as Drosophila. This indicates a complex evolutionary history of gene family evolution by birth-and-death processes quite different from the history of the MT and ER HSP70s but very similar to the patterns seen among the cytosolic heat-inducible HSP70s (Nikolaidis and Nei 2004).
Thomas (2007) recently published an interesting study of P450 genes. In his analysis of complete vertebrate genomes he found that P450s with core functions in development and physiology had very stable phylogenies that reflect organismal relationships; that is, orthologous genes are maintained across great evolutionary distance. However, he reported that among the P450s that function as xenobiotic detoxifiers there were few consistent orthologous relationships and frequent birth-and-death events (Thomas 2007). He suggests that functional liability is then associated with the birth-and-death process. It is well established that both the HSP70s and sHSPs are molecular chaperones. However, the MT and ER HSP70s are constitutively expressed and have crucial roles in the functioning of all cells. The sHSPs have much more varied roles during stress and in some but not all developmental processes. The different patterns of evolution between the HSP70s and sHSPs in C. elegans and C. briggsae agree with the patterns and explanations set forth by Thomas (2007).
In contrast to the evolutionary patterns seen among distantly related species, the pattern of evolution for the sHSPs in the C. elegans and C. briggsae genomes is that of numerous orthologous groups. This is quite similar to the evolutionary patterns seen among the ABC genes (ATP-binding cassette transporters) (Sheps et al. 2004; Zhao et al. 2007). In studies of the ABC genes in C. elegans and C. briggsae, Sheps et al. (2004) also found no orthologous groups across large evolutionary divergence. Further, like the sHSPs, there are orthologous ABC genes in the C. elegans and C. briggsae genomes (Zhao et al. 2007). This pattern of clear sHSP orthology between the C. elegans and C. briggsae genes indicates that these genes originated prior to the split of these two species (approx 80–100 million years ago (Stein et al. 2003). Our analysis identified ten pairs of sHSP C. elegans and C. briggsae orthologs. We also did not identify any singletons in either genome. This indicates that while the deep history of the sHSPs involves birth-and-death, the sHSPs have been very stable in the 80–100 million years since C. elegans and C. briggsae diverged. This is somewhat surprising since a study of the sHSPs in angiosperms found that genomes usually contain novel sHSP genes (Waters et al. in press). More importantly, Stein et al. (2003) noted that there are some gene families in C. elegans and C. briggsae that have expanded in a species-specific manner, most notably the chemosensory receptors. Further, these authors reported that approximately 25% of protein families lack a 1:1 correspondence in the two species. However, we found no evidence of species-specific sHSP gene expansion.
We did find evidence of strong purifying selection among the orthologous C. elegans–C. briggsae pairs. In most cases the Ka for the sHSP orthologs was below (often considerably below) 0.01 and the Ks was near 1.0. Stein et al. (2003) reported that the average Ka between C. elegans and C. briggsae orthologs was 0.011 and the average Ks was 1.78. Therefore many of the sHSPs have a much slower accumulation of amino acid replacements than the average gene. This suggests that high functional constraints have been acting on these genes. We can thus assume that most of the sHSPs have not undergone any functional divergence since the C. elegans and C. briggsae split.
However, based on sequence and gene expression differences it does appear that the orthologous pairs have diverged in function from each other. This conclusion can be drawn from the sequence differences between the homologs; for example, the Ce and Cb 43 kDa proteins are quite distinct from the 17, 20, and 25 kDa proteins. Very little is known about the gene expression patterns for the 17, 20B, and 25 proteins; however, we do know that Ce43 is expressed in the spermathecal tissue, and that Ce20A is crucial for embryonic development (expression data from WormBase). Interestingly, none of these genes are known to be heat-induced. We can then speculate that they are still functioning as chaperones but are no longer important parts of the heat shock response. Because none of these proteins have orthologs in any other species, it is not possible at this time to speculate on how these proteins evolved. Sequences from more divergent nematodes and more gene expression and biochemical data will be helpful in determining how and when these genes evolved in both function and expression.
The four pairs of 12 kDa sHSPs are among the ten orthologous C. elegans and C. briggsae pairs and homologs of these proteins are not known outside of C. elegans and C. briggsae. These proteins are quite interesting because they are missing the N-terminal portion of the sHSP. This region is critical for oligomer formation and chaperone function (van Montfort et al. 2002; Sun and MacRae 2005; Nakamoto and Vigh 2007), and there is compelling evidence that these proteins do not share the highly conserved sHSP oligomer structure or the chaperone function (Leroux et al. 1997a, b; Sun and MacRae 2005). The lack of an N-terminal domain and the ability to be a chaperone strongly suggests a novel protein function or neofunctionalization. The expression patterns for these proteins are also quite interesting. None are known to be heat-induced or to be induced by other stresses (Leroux et al. 1997a, b). But some are known to be present in the spermatocytes. In C. elegans, HSP12.6 is the most highly expressed gene in the dauer-larvae (Jones et al. 2001). This larval state is a type of resting stage that can be induced by stress. What role these proteins have in these tissues is unclear. While we know that the 12 kDa sHSPs are not chaperones at this time we do not know what function they do have. However, the evidence of strong purifying selection acting on these genes in both C. elegans and C. briggsae indicates that their current function is now highly conserved.
Our analysis indicates that the HSP12s and all the other sHSPs are evolving under purifying selection. However, it does appear that the sHSPs may have different functions suggesting that neofunctionalization and positive selection may have one time occurred. The lack of evidence for positive selection among the sHSPs may reflect the difficultly of detecting positive selection when this selection occurred in the distant past and has been followed by purifying selection. It is then possible that the period of positive selection on the sHSPs was so long ago that analysis of current sequences cannot detect it. However, studies of C. elegans and C. Briggsae SRX chemoreceptors (Thomas et al. 2005) and ABC genes (Zhao et al. 2007) did find evidence of positive selection suggesting that if extensive positive selection had been acting on the sHSPs we should have been able to detect it.
There are numerous HSP16 genes in both the C. elegans and C. briggsae genome. The evolution of these genes is quite intriguing. In stark contrast to the other sHSPs in C. elegans and C. briggsae the expression of all of the HSP16 genes are induced by heat shock. However, the HSP16 genes are the only sHSP genes in C. elegans and C. briggsae that do not have clear orthologous relationships. Instead they are found in gene pairs that have very complex evolutionary histories. This unusual gene organization was first identified in C. elegans (Russnak and Candido 1985; Jones et al. 1986). Each gene pair contains two divergent HSP16 genes. One gene pair member is from the I HSP16 lineage and the other from the II lineage. It is clear that individual HSP16 genes are not duplicating by themselves. Instead it is the gene pairs that are duplicating. It is intriguing that all of the C. briggsae HSP16 genes are found in gene pairs but that two of the C. elegans HSP16 gene pairs are not- HSP16A and HSP16B. In addition, both HSP16A and HSP16B are only mildly induced by heat stress. Thus, it appears that only the HSP16s present in gene pairs play a significant role in the heat shock response.
Our analysis shows that the two HSP16 lineages I and II are evolving independently, and it is likely that they have different functions. The Ka values (nonsynonymous substitutions per site) between the I and II genes are roughly 0.50 indicating considerable amino acid divergence between these two types of HSP16s. This in turn suggests that functional differences most likely exist between the two HSP16 lineages. The evidence of purifying selection within each lineage suggests that strong selection exists to maintain both types of HSP16s. There is no evidence of gene conversion homogenizing the HSP16s and removing one or the other lineage. But there is evidence of gene conversion across duplicated pairs indicating that at least some of the gene pairs are being homogenized. That the gene pairs themselves are subject to continuing recombination suggests that there are constraints on functional divergence within each lineage. For example, in C. briggsae there are ten HSP16 genes found in five pairs. Each pair has one type-I HSP16 and one type-II HSP16. It is unlikely that each of these 10 genes is functionally unique. More likely there are five type-I HSP16s with similar or identical functions and five type-II HSP16s that differ functionally from the type-I HSP16s but do not significantly differ from each other. We found no evidence of recent positive selection acting on the HSP16 genes. However, because the gene pairs are present in both C. elegans and C. briggsae we can conclude that the duplication that originally generated these two protein subfamilies predates this organismal divergence and hence occurred more than 100–80 million years ago. Again, it is of course possible that positive selection occurred soon after the original gene duplication that gave rise to the HSP16s and that both gene conversion and strong purifying selection have eroded any evidence of positive selection.
There are a number of other interesting features of the HSP16 gene pairs. One is the presence of snRNA U1 genes. The other is the bi-directional promoter that lies between the two genes (Hong et al. 2004). There are also heat shock elements present in both directions that are needed for expression during heat shock (Hong et al. 2004). However, only one direction (controlling the expression of the type II HSP16 genes) contains a hypoxia response element. Therefore, the type II HSP16 genes but not the type I HSP16 genes are expressed during hypoxia (Hong et al. 2004). The nature of the original HSP16 gene (that is, prior to the first duplication) is not known. But because hypoxia elements are found in both C. elegans and C. briggsae it likely both heat shock and hypoxia response elements were present. It then appears that over time the type I and II HSP16 lineages have diverged in their expression patterns. If the original promoter had both elements, then the loss of expression during hypoxia for some genes would fit the subfunctionalization model of evolution. Under this model the functions or expression patterns of the ancestral gene are subdivided between the two gene duplicates.
Abbreviations
- HSP:
-
Heat shock protein
- sHSP:
-
Small heat shock proteins
- bp:
-
Base pair
- kDa:
-
Kilodaltons
- HSEs:
-
Heat shock response elements
- ESRE:
-
Ethanol stress response element
References
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Boorstein WR, Ziegelhoffer T, Craig EA (1994) Molecular evolution of the HSP70 multigene family. J Mol Evo 38:1–17
Caspers GJ, Leunissen JA, de Jong WW (1995) The expanding small heat-shock protein family, and structure predictions of the conserved “alpha-crystallin domain”. J Mol Evol 40:238–248
de Jong WW, Caspers GJ, Leunissen JA (1998) Genealogy of the alpha-crystallin-small heat-shock protein superfamily. Int J Biol Macromol 22:151–162
Drouin G (2002) Characterization of the gene conversion between the multigene family members of the yeast genome. J Mol Evol 55:14–23
Force A, Lynch M, Pickett FB, Amores A, Yan YL, Postlethwait J (1999) Preservation of duplicate genes by complementary, degenerative mutations. Genetics 151:1531–1545
Franck E, Madsen O, van Rheede T, Ricard G, Huynen MA, de Jong WW (2004) Evolutionary diversity of vertebrate small heat shock proteins. J Mol Evol 59:792–805
Fu X, Jiao W, Chang Z (2006) Phylogenetic and biochemical studies reveal a potential evolutionary origin of small heat shock proteins of animals from bacterial class A. J Mol Evol 62:257–266
Hall T (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows. Nucleic Acids Symp Ser 41:95–98
Haslbeck M, Franzmann T, Weinfurtner D, Buchner J (2005) Some like it hot: the structure and function of small heat-shock proteins. Nat Struct Mol Biol 12:842–846
Higgins DG, Thompson JD, Gibson TJ (1996) Using CLUSTAL for multiple sequence alignments. Methods Enzymol 226:383–402
Hong M, Kwon JY, Shim J, Lee J (2004) Differential hypoxia response of hsp-16 genes in the nematode. J Mol Biol 344:369–381
Jones D, Russnak RH, Kay RJ, Candido EP (1986) Structure, expression, and evolution of a heat shock gene locus in Caenorhabditis elegans that is flanked by repetitive elements. J Biol Chem 261:12006–12015
Jones SJ, Riddle DL, Pouzyrev AT, Velculescu VE, Hillier L, Eddy SR, Stricklin SL, Baillie DL, Waterston R, Marra MA (2001) Changes in gene expression associated with developmental arrest and longevity in Caenorhabditis elegans. Genome Res 11:1346–1352
Kim KK, Kim R, Kim SH (1998) Crystal structure of a small heat-shock protein. Nature 394:595–599
Kumar S, Tamura K, Nei M (2004) MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform 5:150–163
Lee GJ, Vierling E (2000) A small heat shock protein cooperates with heat shock protein 70 systems to reactivate a heat-denatured protein. Plant Physiol 122:189–198
Leroux MR, Ma BJ, Batelier G, Melki R, Candido EP (1997a) Unique structural features of a novel class of small heat shock proteins. J Biol Chem 272:12847–12853
Leroux MR, Melki R, Gordon B, Batelier G, Candido EP (1997b) Structure–function studies on small heat shock protein oligomeric assembly and interaction with unfolded polypeptides. J Biol Chem 272:24646–24656
Lynch M, Force A (2000) The probability of duplicate gene preservation by subfunctionalization. Genetics 154:459–473
Lynch M, Katju V (2004) The altered evolutionary trajectories of gene duplicates. Trends Genet 20:544–549
Mondragon-Palomino M, Gaut BS (2005) Gene conversion and the evolution of three leucine-rich repeat gene families in Arabidopsis thaliana. Mol Biol Evol 22:2444–2456
Nakamoto H, Vigh L (2007) The small heat shock proteins and their clients. Cell Mol Life Sci 64:294–306
Nei M, Rooney AP (2005) Concerted and birth-and-death evolution of multigene families. Annu Rev Genet 39:121–152
Nikolaidis N, Nei M (2004) Concerted and nonconcerted evolution of the Hsp70 gene superfamily in two sibling species of nematodes. Mol Biol Evol 21:498–505
Ohno S (1970) Evolution by gene duplication. New York, Springer-Verlag
Piatigorsky J, Wistow G (1991) The recruitment of crystallins: new functions precede gene duplication. Science 252:1078–1079
Piatigorsky J, Wistow GJ (1989) Enzyme/crystallins: gene sharing as an evolutionary strategy. Cell 57:197–199
Plesofsky-Vig N, Vig J, Brambl R (1992) Phylogeny of the alpha-crystallin-related heat shock proteins. J Mol Evol 35:537–545
Posada D, Buckley T (2004) Model selection and model averaging in phylogenetics: advantages of akaike information criterion and Bayesian approaches over likelihood ratio tests. Syst Biol 53:793–808
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Rost B, Yachdav G, Liu J (2004) The PredictProtein server. Nucleic Acids Res 32:W321–326
Russnak RH, Candido EP (1985) Locus encoding a family of small heat shock genes in Caenorhabditis elegans: two genes duplicated to form a 3.8-kilobase inverted repeat. Mol Cell Biol 5:1268–1278
Sawyer SA (1999) GENECONV: a computer package for the statistical detection of gene conversion. Distributed by the author, Department of Mathematics, Washington University, St. Louis
Sheps JA, Ralph S, Zhao Z, Baillie D, Ling V (2004) The ABC transporter gene family of Caenorhabditis elegans has implications for the evolutionary dynamics of multidrug resistance in eukaryotes. Genome Biol 5:R15
Stein LD, Bao Z, Blasiar D, Blumenthal T, Brent MR, Chen N, Chinwalla A, Clarke L, Clee C, Coghlan A, Coulson A, D’Eustachio P, Fitch DH, Fulton LA, Fulton RE, Griffiths-Jones S, Harris TW, Hillier LW, Kamath R, Kuwabara PE, Mardis ER, Marra MA, Miner TL, Minx P, Mullikin JC, Plumb RW, Rogers J, Schein JE, Sohrmann M, Spieth J, Stajich JE, Wei C, Willey D, Wilson RK, Durbin R, Waterston RH (2003) The genome sequence of Caenorhabditis briggsae: a platform for comparative genomics. PLoS Biol 1:E45
Sun Y, MacRae TH (2005) Small heat shock proteins: molecular structure and chaperone function. Cell Mol Life Sci 62:2460–2476
Thomas JH (2007) Rapid birth-death evolution specific to xenobiotic cytochrome P450 genes in vertebrates. PLoS Genet 3:e67
Thomas JH, Kelley JL, Robertson HM, Ly K, Swanson WJ (2005) Adaptive evolution in the SRZ chemoreceptor families of Caenorhabditis elegans and Caenorhabditis briggsae. Proc Natl Acad Sci USA 102:4476–4481
van Montfort RL, Basha E, Friedrich KL, Slingsby C, Vierling E (2001) Crystal structure and assembly of a eukaryotic small heat shock protein. Nat Struct Biol 8:1025–1030
van Montfort RL, Slingsby C, Vierling E (2002) Structure and function of the small heat shock protein/alpha-crystallin family of molecular chaperones. Adv Protein Chem 59:105–156
Waters ER (1995) The molecular evolution of the small heat shock proteins in plants. Genetics 141:785–795
Waters ER, Vierling E (1999) Chloroplast small heat shock proteins: evidence for atypical evolution of an organelle-localized protein. Proc Natl Acad Sci USA 96:14394–14399
Waters ER, Aevermann BD, Sanders-Reed Z (in press) Comparative analysis of the small heat shock proteins in three angiosperm genomes indentifies new subfamilies and reveals diverse evolutionary patterns. Cell Stress Chaperon
Yang Z (1997) PAML: a program package for phylogenic analysis by maximum likelihood. Comput Appl BioSci 13:555–556
Zhao Z, Thomas JH, Chen N, Sheps JA, Baillie DL (2007) Comparative genomics and adaptive selection of the ATP-binding-cassette gene family in Caenorhabditis species. Genetics 175:1407–1418
Acknowledgements
This manuscript is based on B. D. Aevermann’s Master of Sciences thesis at San Diego State University. Aevermann’s M.S. thesis committee members Drs. R. Zeller and T. Larsen provided useful comments on earlier versions of this manuscript. The Waters lab, and in particular Jim Starrett, provided useful comments and assistance in evaluating the results. We also wish to thank one anonymous reviewer and Dr. R. Krebs for helpful comments on an earlier version of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Aevermann, B.D., Waters, E.R. A comparative genomic analysis of the small heat shock proteins in Caenorhabditis elegans and briggsae . Genetica 133, 307–319 (2008). https://doi.org/10.1007/s10709-007-9215-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10709-007-9215-9