
Abstract
The genetic structure in populations of the Chagas’ disease vector Triatoma infestans from six localities belonging to areas under the same insecticide treatment conditions of Catamarca province (Argentina) was examined at macrogeographical and microgeographical scales. A total of 238 insects were typed for 10 polymorphic microsatellite loci. The average observed and expected heterozygosities ranged from 0.319 to 0.549 and from 0.389 to 0.689, respectively. The present results confirm that populations of T. infestans are highly structured. Spatial genetic structure was detectable at macrogeographical and microgeographical levels. Comparisons of the levels of genetic variability between two temporal samples were carried out to assess the impact of the insecticide treatment. The genetic diversity of the population was not significantly affected after insecticide use since different genetic parameters (allele number, observed and expected heterozygosities) remained stable. However, loss of low frequency alleles and not previously found alleles were detected. The effective population size (N e ) estimated was substantially lower in the second temporal sample than in the first; nevertheless, it is possible that the size of the remnant population after insecticide treatment was still large enough to retain the genetic diversity. Very few individuals did not belong to the local T. infestans populations as determined by assignment analyses, suggesting a low level of immigration in the population. The results of the assignment and first-generation migrant tests suggest male-biased dispersal at microgeographical level.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chagas’ disease (American trypanosomiasis) is a serious public health problem in Latin America, with about 12 million persons infected and around 90 million living in endemic areas (Schmunis 1999). The disease is produced by infection with Trypanosoma cruzi (Chagas), which is transmitted by hematophagous insects of the subfamily Triatominae (Hemiptera: Reduviidae). This subfamily includes more than 130 species, most of which are actual or potential vectors of T. cruzi. Considering the different habitats described for the members of Triatominae, from exclusively sylvatic species to those well adapted to human dwellings, only a few, those with a high degree of adaptation to the domestic environment, have been recognized as effective vectors of trypanosomiasis in humans. Among them, Triatoma infestans (Klug) is one of the species most closely associated with man, widely distributed, and consequently of public health importance. It is the main transmitter of Chagas’ disease in South America between the latitudes 10° S and 46° S, where it is primarily restricted to domestic and peridomestic environments.
The analysis of genetic variability is important in understanding the evolution and dynamics of natural populations. Population analyses based in allozymes of T. infestans produced contradictory results. Dujardin et al. (1987, 1988, 1998) and Pereira et al. (1996) reported low polymorphism inferred from results on two or three polymorphic loci. Contrary to this very low degree of polymorphism, García et al. (1995a, b) detected an important level of genetic variability in laboratory colonies, which could be explained by differences in methodology (García et al. 1995a). Among the newer molecular markers, mitochondrial DNA (mtDNA) genes have been used to analyse T. infestans populations (Monteiro et al. 1999; García et al. 2003). These maternally inherited markers exhibited low levels of polymorphism. As microsatellites or simple sequence repeat markers (Bruford and Wayne 1993; Schlötterer and Pemberton 1994) have permitted greater resolution of genetic variation in populations, 93 microsatellite loci were isolated from partial genomic libraries of T. infestans (García et al. 2004). Previously, we evaluated the possible effect of eradication campaigns on the genetic structure of the vector populations using 10 of these microsatellite loci, comparing levels of genetic variability in natural populations of T. infestans from areas with different elapsed periods since the last insecticide treatment and from areas that never received treatment. Surprisingly, the majority of T. infestans populations from insecticide-treated localities appeared to have retained a substantial proportion of genetic diversity. The existence of subdivision in T. infestans populations could explain this result, since a population bottleneck would result in independent genetic drift effects that could randomly preserve different combinations of alleles in each subpopulation (Pérez de Rosas et al. 2007).
In order to study the macrogeographic and microgeographic genetic structure of T. infestans populations from a region with the same insecticide treatment, here we examine the genetic structure of T. infestans populations using microsatellite markers from six localities belonging to Catamarca province (Argentina), all of which received the last insecticide treatment 5 years before the sample collection. The microgeographical analysis was carried out in one of the localities (Medanitos). With the purpose to assess the impact of the insecticide application, a second sample from Medanitos collected 16 months after insecticide treatment, was also analysed.
Materials and methods
Sampling
The samples studied, their geographical origin (Fig. 1), sample size, and collection dates are shown in Table 1. Samples were collected at four localities of Tinogasta (Medanitos, Saujil, Fiambalá, and Copacabana), one locality in Pomán (Saujil), and one in Capayán (Huillapima) in December of 2004. All these localities received the last insecticide treatment 5 years before the sample collection date. The insects were captured in each place from different peridomiciliary sites, all of which were treated with insecticide after the collection. In Medanitos, the number of individuals collected in each of the three houses sampled (house I, II, and III) ranged from 15 to 31 and the distances between the houses ranged from 140 to 1,220 m. A second sample from Medanitos was collected 16 months later, where only one of those houses, the house III, was found infected with T. infestans (n = 36). A total of 238 individuals of T. infestans obtained from natural populations were independently analysed as described below.

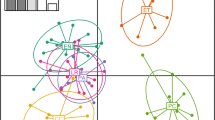
Geographic origin of the Triatoma infestans samples analysed from Catamarca (for more detail see Table 1), and pie chart of combined genetic ancestries of all individuals sampled in each population, as obtained from STRUCTURE (Pritchard et al. 2000). The seven different colors correspond to seven different genetic clusters. Different houses analysed in Medanitos are indicated with numbers I, II, and III
DNA isolation and polymerase chain reaction
The DNA was extracted from legs of each bug using a simplified protocol of a phenol–chloroform extraction procedure reported by Werman et al. (1990). The 10 primer pairs used for polymerase chain reactions (PCR) were previously designed and tested for proper amplification of microsatellite DNA from whole genomic DNA of T. infestans (García et al. 2004). The forward primer from each primer set was 5′-fluorescent labelled with one of three dyes, 6-FAM, HEX, or NED (Applied Biosystems). PCR amplifications were carried out in a Hybaid thermal cycler (Omnigene) in 25 μl of a solution containing 10 mM Tris-HCl (pH 8.3), 50 mM KCl, 1.5 mM MgCl2, each dNTP at 200 μM, each primer at 1 μM, genomic DNA (10–50 ng), and 1 U of Amplitaq Gold (Perkin-Elmer Cetus). Thermal profiles consisted of an initial denaturation step at 94°C for 5 min, followed by 30 cycles of 20 s at 94°C (denaturation), 30 s at 55°C (annealing), and 40 s at 72°C (extension), with a final extension step of 15 min at 72°C. Reaction products were visualized after electrophoresis on a 3% agarose gel (MetaPhor). Size of the PCR products was estimated relative to an internal standard (GeneScan-500 ROX) in polyacrylamide gels using an ABI PRISM 377 automated DNA sequencer, GENESCAN 3.1 and GENOTYPER 2.5 softwares (all from Perkin-Elmer Applied Biosystems).
Analyses
Allelic diversity, and observed (H O ) and expected (H E ) heterozygosities were assessed at each locus. H E was calculated using the unbiased estimate described by Nei (1987). Linkage disequilibrium was tested between all pairs of loci and for all locations using the program FSTAT version 2.9.3 (Goudet 2001). Genotypic disequilibrium was not apparent for any pair of loci according to a global test for each of the 45 different pairs of loci across all populations based on 900 permutations. Therefore, being linkage between loci unlikely, we proceeded under the assumption of statistical independence between loci.
Departures from Hardy-Weinberg (HW) equilibrium were tested by the inbreeding coefficient F IS (Wright 1951) using the estimator f of Weir and Cockerham (1984). The degree of differentiation between and across all sites, and between different houses at one of the localities was determined using Weir and Cockerham (1984) estimator (θ) of Wright’s F ST . On the other hand, Fisher exact test was performed using the software GENEPOP version 3.4 (Raymond and Rouset 1995) to determine allele frequency differences between different houses at one of the localities and between temporal samples.
Single and multilocus f, as well as global and pairwise comparisons of θ were calculated by the program FSTAT version 2.9.3 (Goudet 2001). The significance of all θ and f estimates was tested using permutation. Bonferroni corrections (Rice 1989) for multiple comparisons were applied when necessary. Standard errors of θ were calculated by jackknifing over populations and loci, and a 95% confidence interval was generated by bootstrapping over loci. Significance of global θ estimate was further evaluated with an exact G-test after 1,000 randomizations of allele among sites (Goudet et al. 1996). Isolation by distance was examined by plotting the pairwise θ values against the geographical distances (as straight-line distance between all pairs of sites) and was tested using the Mantel test with permutations as implemented in FSTAT version 2.9.3 (Goudet 2001).
A Bayesian approach implemented in the program STRUCTURE version 2 (Pritchard et al. 2000) was used to infer spatial population structure. The program STRUCTURE uses a Markov chain Monte Carlo (MCMC) approach to infer the number of populations (K) in a data set without prior information of the sampling locations. The no admixture model and uncorrelated allele frequencies within populations were assumed. To choose an appropriate K value for data analysis, a series of eight independent runs for each value of K between 1 and 8 were conducted. In each run, a burn-in period of 100,000 and a 1000,000 run length were performed.
An assignment analysis was performed to identify possible first-generation migrants as implemented by the program GENECLASS2 (Piry et al. 2004). The program uses multilocus genotypes to identify putative immigrants within each population or subpopulation and the most likely source of these immigrants. Moreover, we conducted an assignment test using the exclusion procedure (Bayesian method) to establish whether any individuals could be identified as immigrants from populations or subpopulations other than the sampled. The marginal probability of given individual multilocus genotype was compared to the distribution of marginal probabilities of randomly generated multilocus genotypes (10,000 replicates), and if the P value was <0.01 in case of detection of first-generation migrants, the individual was considered not belonging to the population or subpopulation; when assignment test was carried out, the individual was identified as an immigrant from unsampled population or subpopulation if the P value was <0.01 in all the samples analysed. First-generation migrant and assignment tests were carried out using Rannala and Mountain (1997) Bayesian individual assignment method and the Monte Carlo resampling method of Paetkau et al. (2004).
Two approaches were used to examine any sex differences in immigration and dispersal. On the one hand, contingency table analysis was used to determine whether those individuals identified as putative immigrants (implemented as first-generation migrants by GENECLASS2) differed from a 1:1 sex ratio. On the other hand, the program SPAGeDi version 1.2 (Hardy and Vekemans 2002) was used to determine differences in the mean values of Kinship coefficient (defined as the probability of identity by descent of the genes compared) between males and females within each population or subpopulation. In all cases, it was used the Loiselle et al. (1995) estimator of Kinship coefficient, which is especially suitable in cases when there are low frequency alleles present. The significance of Kinship coefficient estimates was tested using permutation.
Heterozygosity tests compare two estimates of expected heterozygosity, one based on allele frequencies (H E ) and the other based on the number of alleles and sample size (H eq ) (Cornuet and Luikart 1996). If a population experiences a bottleneck, rare alleles will be lost and H eq will decrease faster than H E . This apparent excess of heterozygosity (H E > H eq ) is an indicator of a recent bottleneck event, whereas the converse (H E < H eq ) may indicate an expansion event. Wilcoxon signed-rank tests were used to determine if there was a significant number of loci in which H E > H eq or H E < H eq . Estimates of H eq were calculated under two mutation models, the stepwise mutation model (SMM) and an intermediate two-phase model (TPM) with fractions of mutations greater than one repeat of 10%, 20%, and 30%. We analyse the results based on SMM and TPM models, since the consensus is that they better approximate the mutation process at microsatellite loci than the infinite allele model (IAM) (e.g. Weber and Wong 1993; Di Rienzo et al. 1994; Primmer et al. 1998). Tests were performed on sampling sites and temporal samples using BOTTLENECK version 1.2.02 (Cornuet and Luikart 1996).
Differences in allelic richness (number of alleles corrected for sample size), mean number of alleles, and mean H O and H E values between temporal samples were calculated by Friedman test, followed by Wilcoxon signed-rank tests, using SPSS Base version 11.5.1 (SPSS Inc., Chicago, IL). Linkage disequilibrium method as a one-sample method was used to obtain genetic estimates of the effective population size (N e ) for each temporal sample. This method was applied to the data as implemented in the computer program NeEstimator version 1.3 (Peel et al. 2004). Linkage disequilibrium is the non-random association of alleles at different loci and can be generated by drift in a small population among unlinked loci. This relationship can be exploited to estimate the N e .
Results
Macrogeographical scale
A total of 238 insects from six localities were typed for 10 polymorphic microsatellite loci. Allele frequencies are shown in Appendix I. All loci were polymorphic with the number of distinct alleles per locus ranging from 8 (loci TiC09 and TiE02) to 18 (locus TiC02). The 10 microsatellite loci examined were polymorphic within the six populations investigated with the exception of locus TiC09, which was fixed for allele 139 in the population from Saujil (Tinogasta). Each locus presented exclusive alleles in one of the populations. These private alleles were found in all populations, ranging from one in Saujil (Tinogasta) to 17 in Huillapima (Capayán), and suggest limited current levels of genetic exchange (Pérez de Rosas et al. 2007).
Relatively high genetic diversity levels were detected in all sampled localities (Table 2). The mean number of alleles per locus detected per population ranged from 3.7 in Fiambalá (Tinogasta) to 6.4 in Huillapima. The average observed and expected heterozygosities (H O and H E ) ranged from 0.319 to 0.549 and from 0.389 to 0.689, respectively, with the lowest values in the sample from Fiambalá and the highest values in the sample from Huillapima. Significant departures from Hardy-Weinberg (HW) equilibrium were observed in some of the 10 microsatellite loci in the populations belonging to the six localities studied. Deviations from Hardy-Weinberg expectations were associated with positive F IS indicating excess of homozygotes. Multilocus F IS analyses revealed a significant multilocus heterozygote deficiency in all sampled localities (P < 0.01 and P < 0.001) (Table 2). The most probable causes of the observed heterozygote deficiency could be the population subdivision plus the presence of null alleles (Pérez de Rosas et al. 2007).
Levels of genetic differentiation among the six populations was examined using overall and pairwise comparisons of multilocus F ST (using the estimator θ; Table 3). The overall θ value of 0.169 is significantly different from zero (P = 0.001), as are all pairwise θ values (P < 0.01, FSTAT permutation procedure); θ values ranged from 0.100 between the populations of the geographically closest localities of Saujil and Medanitos (both from Tinogasta) to 0.285 for the comparison between the populations of Fiambalá and Huillapima. There was a significant association between geographical distance and genetic differentiation (θ) among sites (Mantel r = 0.56, P = 0.026), suggesting isolation by distance.
Using the program STRUCTURE version 2 (Pritchard et al. 2000), seven different genetic clusters could be identified (Fig. 1). As suggested by the Bayesian approach, there are six distinct, almost homogeneous clusters distributed across specific, well-separated geographic locations from Catamarca. The samples from Huillapima, Saujil (Pomán), Copacabana (Tinogasta), and Fiambalá form distinct, nearly homogeneous clusters. The geographical closest localities of Medanitos and Saujil (both from Tinogasta) form the two less homogeneous clusters; ∼10% of the ancestry of Saujil is shared with the Medanitos cluster. Another (seventh) cluster was found in low proportion (<30%) in Medanitos and was also found in small proportions (<5%) in both Saujil samples (one from Tinogasta and another from Pomán).
Results of first-generation migrant test for T. infestans from different sampling localities are shown in Table 4. First-generation migrant test allows to identify putative immigrants within each population and the most likely source of this immigrants. Putative migrants were indentified in five populations. Of the nine putative immigrants identified, six connect the populations of the closest localities from Tinogasta (Medanitos, Saujil, and Fiambalá). Five of these individuals were females, three were males, and one was a fifth-instar nymph.
To establish whether any individuals could be identified as immigrants from populations other than the sampled we used the exclusion procedure (Bayesian method). The assignment tests showed that only four T. infestans individuals out of 202 (1.98%) were not assigned correctly to any of the six populations sampled during December 2004. One of these individuals was a male collected in Saujil (Tinogasta) and three of these individuals were females collected in Fiambalá, Medanitos, and Huillapima.
The mean values of Kinship coefficient (defined as the probability of identity by descent of the genes compared) among males and females considered separately in each of the six populations analysed were significant.
Absence of recent bottlenecks was evidenced by heterozygosity tests. Only in one case, the result was dependent on the mutation model. Deviation from mutation-drift equilibrium, as a result of heterozygosity excess, was found in Huillapima under the TPM model with fractions of mutations greater than one repeat of 30%, whereas mutation-drift equilibrium was found when analyses were performed under the SMM and TPM (with fractions of mutations greater than one repeat of 10% and 20%) models. SMM and TPM models, were consistent with heterozygosity deficiency for two of the populations analysed (Saujil and Fiambalá, Tinogasta) and mutation-drift equilibrium for one (Saujil, Pomán), suggesting expansion events in the first case and stability in the second. However, heterozygosity deficiency or stability was detected for Copacabana (Tinogasta) and Medanitos using SMM or TPM models, respectively.
Microgeographical scale
Microgeographical analysis was carried out in Medanitos (Tinogasta), where the number of individuals collected in each of the three houses (I, II, and III) sampled in December 2004 was high enough (from 15 to 31) to analyse them independently. Distribution of allelic frequencies for the microsatellite loci among the insects captured in each of the houses of Medanitos is shown in Appendix I. The level of genetic diversity detected in the different houses was similar (Table 2). Significant deviations from HW expectations observed in some of the loci analysed were associated with excess of homozygotes, with the exception of locus TiE02, which presented a significant excess of heterozygotes in the sample from house I. Multilocus F IS analyses revealed a significant multilocus heterozygote deficiency in samples from two of the three houses analysed in Medanitos (P < 0.01) (Table 2).
Genetic differentiation among the samples obtained in the houses of Medanitos was detected. The overall θ value of 0.095 is significantly different from zero (P = 0.001); F ST estimate significantly greater than zero suggest that there is structuring of allele variance between the three houses. All pairwise θ values were also significantly different from zero (P < 0.01, FSTAT permutation procedure), with θ values ranging from 0.053 to 0.149. In addition, exact tests of allele frequencies revealed significant differences in allele frequencies among the three subpopulations (Table 5). Specifically, houses I–II had significant allele frequency differences at 5 of the 10 loci (50%), houses I–III had significant allele frequency differences at 8 of the 10 loci (80%), and houses II–III had significant allele frequency differences at 5 of the 10 loci (50%).
On the other hand, the Bayesian approach suggested that the T. infestans microsatellite data show the existence of more than one cluster (K) in Medanitos. Two different genetic clusters could be identified in the sample from Medanitos (K = 2, P = 1). The sample from the house I formed a separate cluster. The houses II and III formed another (Fig. 2). However, ∼20% of the ancestry of the house II is shared with the almost homogeneous house I cluster.

Pie chart of combined genetic ancestries of all individuals sampled in each house analysed in Medanitos, as obtained from STRUCTURE (Pritchard et al. 2000). Different houses are indicated with numbers I, II, and III
Putative immigrants were indentified in the three houses of Medanitos using first-generation migrant test. Only one out of 15 (6.67%) and of 24 (4.17%) individuals from houses I and II, respectively, were identified as immigrants (source-house III). In house III, two of 31 (6.41%) individuals were identified as potential immigrants, one from house I and one from house II. Of the four putative immigrants detected, three were males. Contingency table analysis indicated that this was not significantly different from an expected 1:1 sex ratio (χ2 = 0.04, d.f. = 1, P = 0.845).
The assignment tests showed that only five T. infestans individuals out of 106 (4.72%) insects from Medanitos involved in the microgeographical and temporal analysis were not assigned correctly to any of the houses sampled. Three of these individuals were sampled during the first collection date (one male from the house I, one female from the house II, and other male from the house III) and two during the second (two males from the house III). These individuals could be identified as immigrants from subpopulations other than the sampled.
There was not a pattern of differences in the mean values of Kinship coefficient between males and females. These values for males were significant only for both temporal samples from house III, and for females were significant only for house II and second temporal sample from house III (Table 6).
Temporal variation between two samples from Medanitos
To assess the impact of insecticide treatment on the genetic diversity in a population of T. infestans, genetic variability indicators were compared between the first and the second sample, 16 months later, from Medanitos. The average number of alleles (note that allele numbers are not corrected for differences in sample size) was of 6.30 in the first temporal sample and lower (4.90) in the second, although not significantly (P = 0.438). No significant variation was observed between mean H O of the two temporal samples (0.47 and 0.44 for the first and second sample, respectively) as well as between mean H E values for the same samples (0.58 and 0.55 for the first and second sample, respectively). In Medanitos, only one of the three houses sampled in December 2004, the house III, was found infected with T. infestans after insecticide treatment (16 months later). The two temporal samples from house III reveal a higher degree of similarity in the average number of alleles and mean H O and H E (Table 2). However, we detected 11 alleles in the sample collected in house III after insecticide treatment that were not detected in the same house before treatment. Seven of these alleles were also absent in the samples from the other two houses (I and II) sampled in December 2004 (Appendix I). On the other hand, eight alleles present at low frequencies (<5%) in the sample from house III before insecticide treatment were not found in the same house after treatment. Significant differences in allele frequencies were detected for comparison between both temporal samples (Table 5). Allele frequency differences at six of the 10 loci (60%) were detected when all the sample collected before insecticide treatment (from house I, II, and III) was compared with the second temporal sample (from house III); the allele frequency differences were just at three of the 10 loci (30%) when the first and the second sample from the house III were compared.
Changes induced by insecticide treatment on population size have been monitored by estimating the effective population size (N e ) in the population of Medanitos before and after the insecticide treatment. Significant difference in (N e ) estimates, as evidenced by the lack of overlapping of their respective 95% confidence intervals (CI), was detected between the first sample (from house I, II, and III) and the second sample (from house III) of Medanitos, N e = 61 (57–65) and N e = 16 (13–20), respectively. The N e estimates were also significantly different when they were estimated for the first and the second sample obtained in the house III, N e = 100 (49–1,007) and N e = 16 (13–20), respectively. However, heterozygosity tests did not reveal recent bottlenecks and suggested expansion events or stability since heterozygosity deficiency or mutation-drift equilibrium was detected in the second temporal sample from Medanitos, using SMM or TPM models, respectively.
Discussion
Genetic drift and limited gene flow appear to have generated a substantial degree of genetic differentiation among populations of T. infestans (θ = 0.169, P = 0.001) from a region with the same insecticide treatment. Significant isolation by distance, with nearby sites apparently exchanging more genes than distant ones, was also detected. Seven different genetic clusters were identified (Fig. 1). Six populations correspond to six distinct clusters. The population from Medanitos showed higher heterogeneity. Individuals from this locality were found to consist of a mixture of clusters, one of them in a higher proportion (67%) than the seventh cluster identified (29%).
In Medanitos, the genetic diversity of the population was not significantly affected after insecticide treatment since different genetic parameters (allele number, H O and H E ) remained stable. However, we detected either loss of alleles at low frequencies (<5%) in the first sample that were absent in the second or appearance of alleles in the second not found in the first (Appendix I), with significant overall allele frequency differences between both temporal samples (Table 5). One explanation for the absence of diversity loss after insecticide treatment may be the size of the remnant population. The effective population size (N e ) estimated for the population from Medanitos 16 months after insecticide treatment was substantially lower than the N e estimated before that treatment. However, the population had only been studied for two generations after insecticide treatment and the remnant population size probably stayed high enough to maintain its genetic diversity at the pre-treatment level. On the other hand, since the number of generations is limited between the different collections, the appearance of alleles not detected previously to the insecticide treatment would not be consequence of mutations; they may be a result of migration from neighbouring demes, although a low level of immigration was suggested by the presence of very few individuals that did not belong to the local T. infestans populations, as determined by assignment analyses.
Allelic diversity decreases faster than heterozygosity, when a population experiences a reduction of its effective size, and generally develops a heterozygosity excess at neutral loci. The heterozygosity excess persists only a certain number of generations until a new equilibrium is established (Cornuet and Luikart 1996). The results obtained by heterozygosity tests do not support the occurrence of recent bottleneck events either in the populations from the six localities that received the last insecticide treatment 5 years before the sample collection or in the population from Medanitos after 16 months of the first collection and new treatment. Nevertheless, the heterozygosity tests used to detect recent bottleneck events rely on the assumption that each sample is representative of a population with no immigration and no population substructure. If the population is subdivided into several reproductive units, this method may not be able to show evidence of bottlenecks.
In our previous study (Pérez de Rosas et al. 2007), we analysed samples of T. infestans obtained within different houses from Siete Árboles (Chaco province), where there were not peridomestic environments, such as chicken coops and pig or goat corrals, usually invaded by T. infestans. The heterogeneous distribution of allele frequencies suggested the existence of some type of stratification in the population, e.g. subdivision into breeding units with restricted possibilities of genic exchange that may explain the observed heterozygote deficiency (Wahlund effect) (Spiess 1977). The existence of subdivision in T. infestans populations is also strongly confirmed in the present study with specimens collected from peridomiciliary sites in Medanitos (Catamarca province), where specimens within human dwelling were not detected. Significant θ values and significant differences in allele frequencies provide convincing evidence that there is genetic differentiation among subpopulations. Triatoma infestans has been characterized as an insect with restricted dispersal over short distances and usually remains in the same house or in its immediate vicinity during its lifetime (Schofield 1988). In this respect, one should expect mobile species not to show genetic differentiation over very short distances as indicated the results of our microgeographical analysis, unless geographical features and/or very strong selection pressure prevent gene flow.
We tried to identify the actual subpopulations and assign individuals to these subpopulations. Two different genetic clusters were identified in Medanitos. House I shares very low genetic ancestry with houses II and III, therefore, the individuals from house I correspond to a separate cluster and the individuals from houses II and III form another (Fig. 2). The degree of differentiation between houses II and III was the lowest (θ = 0.053, P < 0.01). These houses are the closest, and then genetic interchange is facilitated. In agreement with the results that support the existence of stratification in T. infestans populations, three different clusters were identified in Siete Arboles (Chaco), where samples from four houses were analysed.
A significant excess of homozygotes was observed in the samples from two of the three houses analysed in Medanitos (Table 2). Within two of the four houses analysed in Siete Árboles, there was also a significant excess of homozygotes in some loci (data not shown). This heterozygote deficiency suggests inbreeding and/or a high degree of subdivision in the population. The existence of different randomly mating units (demes) in one house can be tested by considering different regions of the house. We were unable to test a greater stratification within each house because of the limited sample sizes. Investigations on fine-scale genetic structure should be undertaken to clarify this issue.
Potential differences in dispersal strategies by males and females were examined. The assignment tests showed contradictory results at macrogeographical and microgeographical scales. Whereas three females and one male could be identified as immigrants from populations other than the sampled at macrogeographical level, four males and one female could be identified as immigrants from unsampled subpopulations at microgeographical level. In addition, results of first-generation migrant tests showed more males than females (3:1) collected in one house which could come from the other houses sampled at microgeographical level, whereas at macrogeographical range, nine migrants were detected (Table 4), five were females, three were males, and one was a fifth-instar nymph. On the other hand, the mean values of the probability of identity by descent of the genes compared (Kinship coefficient) from males and females within each population were significant indicating strong relationship among all individuals sampled within each locality without evidence of differences in dispersal between both sexes at macrogeographical scale.
Sex-biased dispersal in T. infestans is not well documented, however, relatively short dispersal distances described by both males and females may have a negative impact on the T. infestans ability to colonize new areas. The observed pattern at microgeographical level is consistent with male-biased dispersal. If males are more likely to disperse than females, then levels of Kinship coefficient should be higher among adult females than among adult males within each subpopulation. This is not supported by a pattern of differences in the mean values of Kinship coefficient between males and females at microgeographical level. It is possible that such pattern is not evident due to the small sample size when both sexes are considered separately.
Dispersal may be both active and passive. Passive dispersion of all stages of the insect development and both sexes is principally mediated by man. Even when the levels of immigration were low, the results obtained suggest passive transport at macrogeographical level, whereas active transport, principally through male dispersal, may be possible at microgeographical scale. On the other hand, the appearance of new alleles in the second temporal sample, as probable result of migration from neighbouring demes, does not agree with the significant genetic structuring and low gene flow inferred for populations of T. infestans. Further analyses at microgeographical level must be undertaken to clarify this point and to verify the potential differences in dispersal strategies of males and females.
Abbreviations
- θ:
-
Estimator of Wright’s F ST
- CI:
-
Confidence intervals
- f :
-
Estimator of Wright’s F IS
- F IS :
-
Inbreeding coefficient
- F ST :
-
A measure of the variance of gene frequencies between populations
- H E :
-
Unbiased expected heterozygosity
- H eq :
-
Expected heterozygosity based on the number of alleles and sample size
- H O :
-
Observed heterozygosity
- HW:
-
Hardy-Weinberg
- IAM:
-
Infinite allele model
- MCMC:
-
Markov chain Monte Carlo
- mtDNA:
-
Mitochondrial DNA
- N A :
-
Number of alleles
- N e :
-
Effective population size
- PCR:
-
Polymerase chain reactions
- SMM:
-
Stepwise mutation model
- TPM:
-
Intermediate two-phase model
References
Bruford MW, Wayne RK (1993) Microsatellites and their application to population genetic studies. Curr Opin Genet Dev 3:939–943
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
Di Rienzo A, Peterson AC, Garza JC, Valdes AM, Slatkin M, Freimer NB (1994) Mutational processes of simple-sequence repeat loci in human populations. Proc Natl Acad Sci USA 91:3166–3170
Dujardín JP, Tibayrenc M, Venegas E, Maldonado L, Desjeux P, Ayala FJ (1987) Isozyme evidence of lack of speciation between wild and domestic Triatoma infestans (Heteroptera: Reduviidae) in Bolivia. J Med Entomol 24:40–45
Dujardin JP, La Fuente C, Cardozo L, Tibayrenc M (1988) Dispersing behavior of T. infestans: evidence from a genetical study of field populations in Bolivia. Mem Inst Oswaldo Cruz 83:435–440
Dujardin JP, Schofíeld CJ, Tibayrenc M (1998) Population structure of Andean Triatoma infestans: allozyme frequencies and their epidemiological relevance. Med Vet Entomol 12:20–29
García BA, Soares Barata JM, Blanco A (1995a) Enzyme polymorphism among Triatoma infestans (Hemiptera: Reduviidae) colonies. J Med Entomol 32:126–133
García BA, Canale DM, Blanco A (1995b) Genetic structure of four species of Triatoma (Hemiptera: Reduviidae) from Argentina. J Med Entomol 32:134–137
García BA, Manfredi C, Fichera L, Segura EL (2003) Variation in mitochondrial 12S and 16S ribosomal DNA sequences in natural populations of Triatoma infestans (Hemiptera: Reduviidae). Am J Trop Med Hyg 68:692–694
García BA, Zheng L, Pérez de Rosas AR, Segura EL (2004) Isolation and characterization of polymorphic microsatellite loci in the Chagas’ disease vector Triatoma infestans (Hemiptera: Reduviidae). Mol Ecol Notes 4:568–571
Goudet J (2001) FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3). Available from http://www.unil.ch/izea/softwares/fstat.html. Updated from Goudet (1995)
Goudet J, Raymond M, De Meeus T, Rousset F (1996) Testing differentiation in diploid populations. Genetics 144:1931–1938
Hardy OJ, Vekemans X (2002) SPAGeDi (version 1.2): a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2:618–620
Loiselle BA, Sork VL, Nason J, Graham C (1995) Spatial genetic structure of a tropical understory shrub, Psychotria officinalis (Rubiacea). Am J Bot 82:1420–1425
Monteiro FA, Pérez R, Panzera F, Dujardin JP, Galvão C, Rocha D, Noireau F, Schofield C, Beard CB (1999) Mitochondrial DNA variation of Triatoma infestans populations and its implication on the specific status of T. melanosoma. Mem Int Oswaldo Cruz 94:229–238
Nei M (1987) Molecular Evolutionary Genetics. Columbia University Press, New York
Paetkau D, Slade R, Burden M, Estoup A (2004) Genetic assignment methods for the direct, real-time estimation of migration rate: a simulation-based exploration of accuracy and power. Mol Ecol 13:55–65
Peel D, Ovenden JR, Peel SL (2004) NeEstimator: software for estimating effective population size. Version 1.3. Queensland Government. Department of Primary Industries and Fisheries
Pereira J, Dujardin JP, Salvatella R, Tibayrenc M (1996) Enzymatic variability and phylogenetic relatedness among Triatoma infestans, T. platensis, T. delpontei and T. rubrovaria. Heredity 77:47–54
Pérez de Rosas AR, Segura EL, García BA (2007) Microsatellite analysis of genetic structure in natural Triatoma infestans (Hemiptera: Reduviidae) populations from Argentina: its implication in assessing the effectiveness of Chagas’ disease vector control programmes. Mol Ecol 16:1401–1412
Piry S, Alapetite A, Cornuet J-M, Paetkau D, Baudouin L, Estoup A (2004) GENECLASS2: a software for genetic assignment and first-generation migrant detection. J Hered 95:536–539
Primmer CR, Saino N, Moller AP, Ellegren H (1998) Unraveling the process of microsatellite evolution through analysis of germ line mutations in barn swallows Hirundo rustica. Mol Biol Evol 15:1047–1054
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Rannala B, Mountain JL (1997) Detecting immigration by using multilocus genotypes. Proc Natl Acad Sci USA 94:9197–9201
Raymond M, Rousset F (1995) GENEPOP (versión 1.2): population genetics software for exact tests and ecumenicism. J Hered 86:248–249
Rice WR (1989) Analysing tables of statistical tests. Evolution 43:223–225
Schlötterer C, Pemberton J (1994) The use of microsatellites for genetic analysis of natural populations. In: Schierwater B, Streit B, Wagner GP, DeSalle R (eds) Molecular ecology and evolution: approaches and applications. Birkhauser Verlag, Basel, pp 203–214
Schmunis GA (1999) Iniciativa del Cono Sur. In: Proceedings of the 2nd international workshop on population biology and control of Triatominae, Santo Domingo de los Colorados, Ecuador, INDRE, Mexico City, pp 26–31
Schofield CJ (1988) Biosystematics of the Triatominae. In: Service MW (ed) Biosystematics of haematophagous insects, systematics association special, Vol 37. Clarendon, Oxford, pp 285–312
Spiess EB (1977) Genes in populations. John Wiley & Sons, New York
Weber JL, Wong C (1993) Mutation of human short tandem repeats. Hum Mol Genet 2:1123–1128
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Werman SD, Davidson EH, Britten RJ (1990) Rapid evolution in a fraction of the Drosophila nuclear genome. J Mol Evol 30:281–289
Wright S (1951) The genetical structure of populations. Ann Eugen 15:323–354
Acknowledgments
We thank Dr. Antonio Blanco for critical revision of the manuscript and Octavio Fusco for technical assistance. We also thank Blanca Herrera de Bravo, Arturo Herrera, and coworkers of the National Programme of Chagas Control-Catamarca. Research was supported by the grants from the UNDP/World Bank/WHO Special Programme for Research and Training in Tropical Diseases (TDR), the Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) of Argentina, and the Secretaría de Ciencia y Tecnología de la Universidad Nacional de Córdoba. A. R. Pérez de Rosas is a Fellow, and E. L. Segura, L. Fichera, and B. A. García are Career Investigators of CONICET.
Author information
Authors and Affiliations
Corresponding author
Appendix
Appendix
Rights and permissions
About this article
Cite this article
Pérez de Rosas, A.R., Segura, E.L., Fichera, L. et al. Macrogeographic and microgeographic genetic structure of the Chagas’ disease vector Triatoma infestans (Hemiptera: Reduviidae) from Catamarca, Argentina. Genetica 133, 247–260 (2008). https://doi.org/10.1007/s10709-007-9208-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10709-007-9208-8