
Abstract
Soil microbes are essential links between above- and below-ground ecosystems and play an important role in regulating ecological functions in the soil. Dynamic interactions within the soil-microbial community in a cereal-legume intercropping ecosystem influence the composition and structure of N-cycling microbial groups (e.g. nitrogen-fixing bacteria). However, these effects have not been extensively studied in some intercropping patterns or in response to varying nitrogen fertilization levels. In the present study, we evaluated the effects of reduced and conventional nitrogen application in a sweet maize (Zea may L.)/soybean (Glycine max L.) strip intercropping system under three cropping patterns over a 3-year time period. High-throughput sequencing and quantitative PCR techniques were used to investigate changes to both the microbial community structure and the expression of key nitrogen-cycling genes in the rhizosphere. Our results indicate that reduced nitrogen application affected the microbial community structure in the rhizosphere, but microbial diversity in the sweet maize rhizosphere was relatively stable. Both the abundance and activity of functional marker genes for microbial nitrogen fixation (nifH), nitrification (amoA), denitrification (nirS, nirK, nosZ), and decomposition (chiA) increased significantly from 2013 to 2016. Taken together, these data demonstrate that the quantified shifts in the soil microbial community and the observed increases in the expression of key functional genes involved in N-cycling were the result of reduced nitrogen application in this strip intercropping system. This study, therefore, provides essential insight into the potential relationships between functional nitrogen-cycling genes and mitigation of nitrogen-loss and N2O emissions in a cereal-legume strip intercropping system.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Cereal-legume strip intercropping systems are composed of two or more crops species that coexist on the same field at the same time, it is a potential practise for crop intensification in both time and space dimensions by complementary resource utilization among cereals and legumes species (Brooker et al. 2015; Zhu et al. 2015). In this environment, the dynamic interactions between the roots and rhizosphere (e.g. sensing, signalling, and acquisition of nutrients) will influence the physiology of the whole plant (Li et al. 2016) and will also influence the composition, structure, and activity of soil microorganisms (Cong et al. 2015; Duchene et al. 2017). These microorganisms are known to play an important role in nutrient transformation and cycling in strip intercropping ecosystems (Li et al. 2014). Furthermore, the diversity of the microbial community structure in the rhizosphere soil of strip intercropping systems is generally higher than that found in monocropping systems (Bainard et al. 2013). Moreover, cereal-legume strip intercropping has been shown to further alter the composition and structure of the functional microbial community, especially with regards to the organisms involved in soil nitrogen transformation processes in the rhizosphere, such as nitrogen (N) fixation, ammonia-oxidation, (de)nitrification, nitrous oxide reduction, and organic N decomposition (Hai et al. 2009). It was also recently demonstrated that strip intercropping systems contribute to soil N sequestration by increasing biological N fixation and/or reducing greenhouse gaseous N loss. For instance, root exudates from maize stimulate nodulation and symbiotic N2 fixation in the rhizosphere of faba bean (Vicia faba L.) when they are intercropped with maize (Zea may L.) (Li et al. 2016). Further, in a wheat (Triticum aestium L.)/soybean (Glycine max L.) intercropping system, it was shown that intercropping changed the dominant species of ammonia-oxidizing bacteria, affected the composition and structure of the soil microorganisms, and increased functional genes expression (e.g. amoA, nirK, nirS, and nosZ) in the rhizosphere (Wallenstein et al. 2006). Intercropping, however, is not the only factor affecting the rhizosphere.
The amount of N fertiliser applied to the plants is another known factor that affects microbial communities involved in N-cycling and transformation. For instance, the abundance of Proteobacteria and Bacteroidetes increased, while that of Acidobacteria decreased, in the high-N plots in long-term N-application experiments (Fierer et al. 2012). High and low rates of ammonium fertiliser applied in agricultural fields were also observed to change the composition and functional traits of ammonia-oxidizing bacteria (AOB) and archaea (AOA). At a continuously low N fertiliser level, AOA become more dominant in ammonium oxidation processes and yield less N2O emission compared to AOB (Hink et al. 2018). Our previous study demonstrated that sweet maize/soybean strip intercropping with reduced N fertiliser could lower soil mineral N loss and mitigate cumulative soil N2O emissions in tropical and subtropical regions (Tang et al. 2017). Thus, the integration of soil microbial community and cropping management can provide insights into N-cycling and transformation in agricultural ecosystems and further link the soil microbial community and environmental changes (Xue et al. 2013). However, the mechanisms underlying the effects of strip intercropping with reduced N fertiliser on the soil microbial community and N-cycling in intensive farming system are not well understood.
In this study, high-throughput sequencing (Illumina MiSeq) and quantitative PCR techniques were applied to reveal the effects of N fertiliser reduction and intercropping patterns on the soil bacterial community structure and on functional microbial groups involved in key processes of nitrogen transformation. Experiments were conducted using varying cropping patterns and two N fertiliser application rates in a sweet maize/soybean strip intercropping system and the structure and composition of the soil microbial community was evaluated. Furthermore, the expression of key functional genes involved in N-cycling were also investigated in an intercropping system with soybean over the course of 3-years of reduced nitrogen application. To our knowledge, this is the first time the changes in composition, structure, and functional gene abundance of soil microbial communities under different cropping patterns and N application rates have been evaluated.
Materials and methods
Experimental site and design
A long-term field experiment, initiated in 2013 and continued to 2016, was carried out at South China Agricultural University (23°8′N, 113°15′E). Detailed information concerning the experimental site and crop management in the sweet maize field are described in Tang et al. (2017). The experimental design was a two-factor randomized complete block. Factor A was cropping system and factor B was N fertilization rate. Briefly, two different N fertiliser rates were applied to the sweet maize field at a reduced rate (300 kg N ha−1, designated N1) and a conventional rate (360 kg N ha−1, designated N2). The cropping patterns were sweet maize in solo-culture (SS) as well as sweet maize-soybean intercropping with crop line ratios of 2:3 (S2B3) and 2:4 (S2B4). S2B3 indicates two rows of sweet maize intercropping with three rows of soybean, while S2B4 indicates two rows of sweet maize with four rows of soybean. In total, there were six cropping pattern combinations and fertiliser rates investigated in this study: SS-N1, SS-N2, S2B3-N1, S2B3-N2, S2B4-N1, and S2B4-N2. Each treatment arranged one plot in three blocks [repeated three times, (n = 3)], and therefore generating 18 plots in total.
Soil sample collection and chemical analyses
Soil samples were collected by auger (0.08 m in diameter) at a soil depth of 0–0.2 m when the crops were ripe. In the intercrop plots, two cores were collected from sweet maize rows and two cores from soybean rows mixed thoroughly to represent one replicate for one treatment. In the sole crops, four soil cores were collected from each plot and mixed thoroughly to represent one replicate. Totally, 18 mixed soil samples per year (three replicates per treatment) were used for soil chemical property and nutrient content determination. The following analyses were conducted according to the methods established by Bao (2013): soil organic matter (SOM) was detected with an oil bath-heated potassium dichromate oxidation reaction, total nitrogen (TN) was detected by semi-micro Kjeldahl, total phosphorus (TP) was detected by molybdenum-antimony colorimetry, total potassium (TK) was detected by flame photometry, available N (AN) was detected by steam distillation, available P (AP) was detected by colorimetry, and available K (AK) was detected by the atomic absorption spectrometry. Soil nitrate nitrogen (NO3−) and ammonium nitrogen (NH4+) were detected by cadmium reduction via a continuous flow analyzer (AA3HR, SEAL, Germany).
Rhizosphere soil sampling was carried out from six treatments in the autumn of 2013 and 2016 when the sweet maize was ripe. In the sole plots, the sweet maize was sampled randomly, whereas in the intercropping plots, only sweet maize neighbouring soybean plants were sampled. In each plot, five plants were excavated, and the loosely adhering soil was shaken off. The tightly adhered rhizosphere soil was collected from the roots and pooled into one mixed rhizosphere soil sample per plot. In total, 36 rhizosphere soil samples were collected and stored immediately at 4 °C, followed by transfer to − 20 °C until further analysis.
Soil DNA preparation and microbial diversity analysis
Soil DNA was extracted from the rhizosphere soil samples using a Fast DNA SPIN KIT (Q·BIOgene) according to manufacturer’s protocol. DNA concentration and purity were determined with a NanoDrop 2000 UV–Vis spectrophotometer (Thermo Scientific, Wilmington, USA), and DNA quality was checked by 1% agarose gel electrophoresis.
The V3–V4 hypervariable regions of the bacteria 16S rRNA gene were amplified with primers (338F: 5′-ACTCCTACGGGAGGCAGCAG-3′ and 806R: 5′-GGACTACHVGGGTWTCTAAT-3′) using a thermocycler PCR system (GeneAmp 9700, ABI, USA). The PCR reactions were conducted using the following program: 3 min of denaturation at 95 °C, followed by 27 cycles of 30 s at 95 °C, 30 s at 55 °C, and 45 s at 72 °C, and a final extension at 72 °C for 10 min. PCR reactions were performed in triplicate using 20 μL mixtures containing 4 μL of 5X FastPfu Buffer, 2 μL of 2.5 mM dNTPs, 0.8 μL of each primer (5 μM), 0.4 μL of FastPfu Polymerase, and 10 ng of template DNA. The PCR products were extracted from a 2% agarose gel and further purified using an AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA). The products were quantified using QuantiFluor™-ST (Promega, USA) according to the manufacturer’s protocol.
Sequencing was performed by Majorbio Bio-Pharm Technology Co., Ltd. (Shanghai, China). The raw reads were deposited into the NCBI Sequence Read Archive (SRA) database (No. SRP148714). The purified amplicons were pooled using equimolar amounts and paired-end sequenced (2 × 300) on an Illumina MiSeq platform (Illumina, San Diego, USA) according to standard protocols. Forward and reverse sequences were merged by overlapping paired-end reads using FLASH with a required overlap length of 10–100 bp. For all 36 samples, the average merged read length was 463 bp, and then the sequence reads were further quality filtered using software Trimmomatic. Briefly, the index sequences contained in the first 12 bp of each paired-end read were extracted and concatenated to form a 24 bp dual-index barcode specific for each paired read and sample. Additional sequence reads pre-processing included: (1) removal of primer sequence; (2) sequence reads were truncated at any site or received an average quality score < 20 over a 50 bp sliding window and (3) removal of trimmed reads having less than 75% of their original length, as well as its paired read (Fadrosh et al. 2014). Operational taxonomic units (OTUs) were clustered at a 97% similarity cut-off using UPARSE (version 7.1 http://drive5.com/uparse/). The taxonomy of each 16S rRNA gene sequence was analysed by RDP Classifier algorithm (http://rdp.cme.msu.edu/) against the Silva (SSU123) 16S rRNA database using a confidence threshold of 70%.
Real-time quantitative PCR assay
Real-time quantitative PCR (qPCR) was conducted on a 7500 real time PCR system (Applied Biosystems, Germany) to quantify the expression of nitrogen fixing (nifH), ammonia-oxidizing (amoA), denitrifying (nirK and nirS), nitrous oxide reducing (nosZ), and organic nitrogen decomposing (chiA) genes in the rhizosphere soil samples, primer sets for each gene listed in Supplementary Table 2. The SYBR Premix PCR kit (TaKaRa Biotechnology, Otsu, Shiga, Japan) used to determine the amount of target DNA in the initial sample. Each reaction was performed in a 20 μL volume containing 1 μL of a tenfold dilution of DNA template (1–10 ng), 10 μL of SYBR Premix Ex Taq, 0.4 μL of each primer (10 μmol L−1), and 8.2 μL of sterile double distilled water (Williamson et al. 2000).
Melting curve analysis was performed at the end of each real-time PCR run to check the amplification product specificity before confirmation by standard agarose gel electrophoresis. The program used for melting curve generation was: 95 °C for 15 s, 60 °C for 15 s, and 95 °C for 15 s. Standard curves of known copy numbers of each gene were generated by following procedures. The reference amplicon was generated from purified PCR products, which amplified from genomic DNA extracted from soil samples using each primer pair. The PCR products and then cloned into the pMD19-T cloning vector. Plasmids containing the correct insert (determined by sequencing) were then expressed and harvested from recombinant Escherichia coli. Plasmid DNA was extracted, and its concentration (ng μL−1) was measured spectrophotometrically using a NanoDrop® ND-1000 spectrophotometer (NanoDrop Technologies). Using a tenfold serial dilution of each plasmid DNA, a standard curve for each target gene was constructed by plotting Ct as a log-based function of the copy number using the following formula: logT0 = –logE × Ct + logK, where E is the PCR efficiency, T0 is the initial amount of DNA, and K is the calculated initial amount of DNA for a Ct value of 0.
Statistical analysis
An α-diversity analysis of the microbial community for each treatment was conducted using the Chao and Shannon indices based on the Illumina MiSeq sequencing data. Comparisons between treatments were performed by a one-way ANOVA using the SPSS 19.0 software package. Mean values (n = 3) among treatments and years were compared with the least significant difference (LSD) by Duncan’s multiple range tests at a 5% significance level.
The relationships between the relative abundance of specific microbial taxa and soil chemical properties were examined by redundancy analysis (RDA). Non-metric multidimensional scaling (NMDS) was also conducted to visualize patterns in the soil microbial community composition and crop management parameters. Additionally, dissimilarity test, analysis of similarity (ANOSIM), and multiple response permutation procedure (MRPP) algorithms were used to test the significant differences among different treatments. All statistical analyses were performed in R v.3.3.1 using the “vegan” package.
Results
Effects of intercropping and N fertiliser treatment on soil chemical properties
Our analysis shows that many of the chemical properties and fertility parameters of the soil, including SOM, TN, TP, TK, AN, NH4+, and NO3− levels, were similar for all six treatments within the first growing season in both 2013 and 2016 (Table 1). However, we did observe that the pH, available P, and available K were affected over the course of the 3-year experiment. Indeed, the pH was significantly lower for the SS-N12, SS-N2, S2B3-N1, S2B4-N1, and S2B4-N2 treatments in 2016 than it was for these treatments in 2013. Further, available P was significantly increased in the S2B4-N1 and S2B4-N2 treatments in 2016 compared to the levels observed for these treatments in 2013. Available K was also significantly increased in the S2B4-N2 treatment in 2016 compared to the levels observed in 2013.
α-Diversity distribution of microbial communities in the sweet maize rhizosphere
Soil DNA samples from the sweet maize rhizosphere (18 from 2013, 18 from 2016) were analysed by two-terminal sequencing. In total, 4,089,810 raw sequences were generated that contained 1,353,289 effective sequences. The average sequence length was 436.47 bp and accounted for 99.98% of the total sequences. According to the similarity of the sequences, the effective sequences were classified into 3253 OTUs. Soil microbial diversity and species classification were determined for the OTUs using the Silva (Release115) 16S rRNA database at a 97% nucleotide sequence identity threshold. The results of this sequence analysis indicate that the 3253 OTUs include 34 phyla, 664 families, and 1304 species. The rarefaction curves were based on evenly rarefied OTUs abundance and observed richness as Sobs index (Supplementary Fig. 1) shows that the 36 DNA samples tended to reach the plateau stage, indicating that the sequencing data are reasonable and reliable.
A species Venn diagram analysis revealed 33 and 89 unique OTUs in 2013 and 2016, respectively, indicated that about 96.2% (3131 out of 3253) of the OTUs were shared by both growing seasons. Furthermore, bacterial community analysis of the 36 soil samples indicated no significant differences in microbial richness and diversity for the Chao, Shannon, and Evenness indices with regards to cropping or planting duration (Table 2). However, the Chao index was significantly lower in the high fertilization (conventional) treatments SS-N2 and S2B3-N2 and higher in the reduced fertilization treatment S2B3-N1 in 2013 compared to that calculated for these treatments in 2016. There were no significant differences in the Chao index among the other treatments between 2013 and 2016. The Shannon index was significantly higher for the reduced fertilization treatments S2B3-N1 and S2B4-N1 than that of the conventional fertilization treatment S2B3-N2 in 2016, but there was no significant difference among all treatments in 2013 compared to the equivalent treatment in 2016. The Evenness’s index was significantly higher for the S2B4-N1 treatment in 2016 than that calculated for this treatment in 2013, but no significant differences were observed among the other treatments. Thus, this analysis indicates that while the mode of cropping and planting duration had no significant effect on the microbial α-diversity in the sweet maize rhizosphere soil in the mature stage, there were no apparent trends in microbial richness and diversity under the two levels of nitrogen fertiliser in all six treatments. In general, microbial diversity in the sweet maize rhizosphere was relatively stable over our 3-year experiment.
Soil microbial diversity and community composition at the phylum and family levels in the sweet maize rhizosphere
The relative abundance of the five most dominant bacterial groups at the phylum level are shown in Table 3. The distribution of the bacterial groups in the sweet maize rhizosphere was similar in 2013 and 2016. The community was composed of 12 phyla and the five most abundant across all six treatments, which account for 80% of the total sequences, were Proteobacteria, Actinobacteria, Chloroflexi, Acidobacteria, and Bacteroidetes. There were no significant differences in the Proteobacteria, Actinobacteria, Chloroflexi, or Bacteroidetes classes among the treatments in 2013, but there were significant (P < 0.05) treatment-dependent differences in Acidobacteria (Table 3). In 2016, there were no significant differences among the six treatments for Actinobacteria, Chloroflexi, or Acidobacteria, but the abundance of Proteobacteria and Bacteroidetes did vary depending on treatment (P < 0.05). A three-way ANOVA revealed that while the cropping patterns and fertiliser rates did not affect the relative abundance of the five most abundant bacterial communities at phylum level, the abundance of Actinomycetes and Chloroflexi varied significantly over the course of the 3-year experiment (P < 0.05) (data not shown).
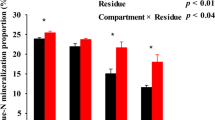
At family level, the dominant bacterial groups were identical between 2013 and 2016 (Fig. 1a). The most abundant bacterial group was Sphingomonadaceae, followed by Xanthomonadaceae, Burkholderiaceae, Chitinophagaceae, Streptomycesceae, Norank_p_Saccharibacterial, Gemmatimonaceae, Bacillaceae, and Rhizobiaceae. In 2013, the relative abundance of Sphingomonadaceae varied depending on the fertiliser rate in the S2B3 cropping pattern, with a significantly higher amount being observed for the reduced fertilization treatment than for the conventional fertilization treatment. Similarly, the relative abundance of Bacillaceae and Rhizobiaceae in the 2016 S2B3 cropping pattern was also dependent on fertiliser rate. Indeed, several of the other bacterial groups varied with cropping pattern and fertiliser rate as shown in Fig. 1b. For example, Xanthomonadaceae was higher in the conventionally fertilized SS and S2B3 cropping patterns, but Chitinophagaceae and Bacillaceae were higher in the reduced fertiliser treatment of the SS cropping pattern. Further, Burkholderiaceae also changed significantly in the S2B3 cropping pattern, being more abundant in the reduced fertilization treatment than it was in the conventional fertilization treatment.

a, b Relative abundance of dominant bacterial groups in six different treatments of 2013 and 2016 (%). Values are mean ± SE; Asterisk above the values of autumn of 2016 indicated a significant difference (*P < 0.05, **P < 0.01) of relative abundance in the same treatment but in different growing year (t test)
Distribution of microbial community β-diversity in the sweet maize rhizosphere
A non-parametric multivariate ANOVA test and an NMDS were conducted to evaluate the variation in the microbial communities caused by cropping patterns and fertilization levels (Fig. 2). The NMDS analysis of bacterial community structure in 2016 and the non-parametric multivariate ANOVA showed that nitrogen application rate had a significant effect on the bacterial community structure. Furthermore, there were significant differences between 2013 and 2016, indicating that the time of planting also had an important effect on the bacterial community structure. In 2016, 18 soil samples were also observed to be more similar than the samples collected in 2013. Notably, the results of these analyses were verified using the ANOSIM and MRPP algorithms.

NMDS analysis of soil bacteria community structure in sweet maize rhizosphere in 2013 and 2016. Similarities or differences among sampling times and treatments are indicated by symbols and letters, respectively, as described in the legend
Linking soil microbial community structure and soil chemical properties
The relationships between the relative abundance of specific microbial taxa and the soil chemical properties were examined by detrended correspondence analysis (DCA) of the sequencing data. The maximum value of the first four axes was 1.45, and a linear model RDA was used to evaluate the interrelationships. Cumulatively, the known environmental variables explained 35.87% of the variation in microbial communities among samples (Fig. 3). Their order of influence was: pH > NH4+ > AP > TK > NO3− > SOM > AK > TN > AN > TP. Based on the RDA model, the first axis explained 28.89% of the variations in bacterial community environmental parameters, while the second axis explained only 6.98% (Fig. 3). The bacterial communities in 2013 and 2016 were then tested separately and, according to the RDA analysis (Supplementary Table 1), soil pH (P < 0.01, r = 0.61), NH4+ concentration (P < 0.01, r = 0.5), available P (P < 0.01, r = 0.37), and total K (P < 0.05, r = 0.22) were the four most important contributors to the variation in bacterial communities. Thus, soil with similar pH, NH4+ concentration, available P, or total K tended to harbour similar bacterial communities.

The RDA of sequence data under three cropping patterns and two nitrogen rates with selected environmental variables for soil bacterial community structure in sweet maize rhizosphere. Similarities and differences among sampling times and treatments are indicated by symbols and letters above symbols, respectively, as described in the legend. The selected variables included soil organic matter (SOM), total nitrogen (TN), total phosphorus (TP), total potassium (TK), available N (AN), available P (AP), available K (AK), nitrate nitrogen (NO3−), ammonium nitrogen (NH4+)
qPCR analysis of key functional genes associated with nitrogen cycling bacteria
In contrast to 2013, the expression levels of six N-cycling functional marker genes were significantly increased in 2016 (Table 4). The abundance of AOB was quantified based on amoA gene copy number, which increased by 4.16-fold/g of dry soil in all six treatments from 2013 to 2016. However, no significant differences were observed for the six treatments within the same year. The abundance of N-fixing bacteria was determined based on nifH gene copy number. nifH expression increased by at least 29.1-fold (in S2B4-N2) and up to 70-fold (in S2B3-N1) from 2013 to 2016. Furthermore, while fertilization level did not influence nifH copy number in 2013, expression was significantly lower in the conventional fertilization treatments (S2B3-N2 and S2B4-N2) than it was for the reduced fertilization treatments (S2B3-N1 and S2B4-N1) in 2016. Nitrite-reducing bacteria were quantified by determining the copy numbers of nirK and nirS. nirK copy number/g dry soil increased over the course of the experiment, ranging from 7.5- (S2B3-N2) to 55.9-fold (S2B3-N1), but no significant differences were observed among the six treatments within 2013 or 2016. nirS expression also increased from 2013 to 2016 (maximum: 6.9-fold in S2B4-N1, minimum: 1.6-fold in SS-N2). Similar to nifH, the N fertilization rates appear to play a significant role in determining nirS copy number in the S2B3 and S2B4 cropping patterns, with the reduced fertilization treatments having higher nirS expression than the conventional fertilized treatments. Moreover, nosZ gene copy number was used to evaluate the abundance of nitrous oxide-reducing bacteria. nosZ expression initially increased, ranging from 12.8-fold in S2B3-N1 to 25.7-fold in S2B4-N1. While there was no significant differences in expression among the six treatments within the same year, nosZ gene copy number was significantly increased in 2016 compared to that observed in 2013. Finally, the chiA gene is often used as a molecular marker of organic nitrogen regulation in soil. In this study, there was no significant difference in the expression of this gene among the six treatments in 2013 or 2016. However, gene copy number varied greatly, ranging from 19.1-fold in SS-N2 to 51.3-fold in S2B4-N1, over the course of the experiment.
A three-way ANOVA of the expression of all six functional N-cycling genes in the sweet maize rhizosphere showed that cropping system and N fertiliser rate had no remarkable effect on amoA, nirK, nirS, nosZ, and chiA genes, but the cropping year significantly affected the expression of all of these genes (P < 0.01, Table 5). Notably, expression of the N fixing gene nifH was affected by cropping system and N fertilization as well as the cropping year (P < 0.01). An RDA of functional gene expression and the soil bacterial community structure showed that 25.3% of the total variance in the soil bacteria community was covered by axes I and II (21.12% and 3.91%, respectively; Supplementary Fig. 2). Correlation analysis also showed that bacterial community structure was closely related to amoA (P < 0.01), nirK (P < 0.01), nirS (P < 0.05), and nosZ (P < 0.01) expression, indicating that bacterial community structure would likely have a significant effect on the nitrification, denitrification, and N fixation processes in the soil.
Discussion
Understanding changes to the composition and structure of soil microorganisms in response to fertilization rate over time is important for maintaining a sustainable intercropping system (Martin-Guay et al. 2018; Gaba et al. 2015; Philippot et al. 2013). In the present study, we evaluated the changes occurring in the microbial community structure and in the expression of six key functional N-cycling genes in the rhizosphere of sweet maize under different strip intercropping combinations and different N application rates. Our results showed that the composition and structure of the microbial community were markedly modified over the course of this 3-year experiment, and the six key functional genes involved in N-cycling were also highly enriched during this time. To our knowledge, this is the first time these changes in composition, structure, and functional gene abundance have been evaluated for sweet maize grown in tropical and subtropical regions under these particular conditions.
Notably, the α-diversity indices show that there was no significant effect of cropping pattern or N application rate on bacterial community composition and structure, suggesting that the geographical characteristics and soil bacterial community composition and structure were relatively stable. This may be due to the short duration of the field trial (3 years) which may not fully demonstrate the effects of these on the bacterial community (Fan et al. 2012). Furthermore, previous research also indicates that soil bacteria may be more influenced by the geographical characteristics of the soil (e.g. forest land vs. grassland vs. farmland) than they are by crop management in an agriculture ecosystem (Lauber et al. 2008).
In our analysis, Proteobacteria, Actinobacteria, Chloroflexi, Acidobacteria, and Bacteroidetes were the most dominant phyla in the rhizosphere of sweet maize, which was similar to the results of a previous study investigating bacterial diversity in the maize rhizosphere (Correa-Galeote et al. 2016). Interestingly, Actinomycetes is known to promote the decay of plant and animal residues in the soil and plays an important role in the decomposition of organic matter after straw returning, highlighting its involvement in the N cycle of the soil ecosystem (Lauber et al. 2008). Compared to 2013, the relative abundance of both Actinobacteria and Chloroflexi decreased in 2016, which may be explained by changes in the soil chemical properties due to continuous fertilization (Shi et al. 2015).
At the family level, there were no changes in the abundance of Chitinophagaceae, Xanthomonadaceae, Bacillaceae, Norank_p_Saccharibacterial, Streptomycetaceae, or Gemmatimonadaceae among the different cropping patterns or fertiliser rates. It has been suggested that these taxa might be more common colonizers than Sphingomonadaceae, Burkholderiaceae, and Rhizobiaceae in the sweet maize/soybean intercropping rhizosphere (Hargreaves et al. 2015). However, the abundance of soil bacterial groups at phylum- or family-level do not always reveal that functional features in sites, more research work should operate at species level in situ in future.
It is important to note that soil microbial community composition is significantly correlated with changes in soil chemical properties (Lauber et al. 2008; Campbell et al. 2010). Indeed, the pH of the soil significantly decreased in all treatments over the 3-year experiment, which may be due to continued N fertilization (especially NH4) of the soil and/or the release of H+ into the soil by nitrification. Our results are similar to those of previous studies showing that continuous application of fertiliser reduces soil pH (Shen et al. 2010; Berthrong et al. 2014). Thus, it is likely that the different fertilization rates applied to the sweet maize/soybean strip intercropping system shaped the bacterial community structure via changes in the chemical properties of the soil. This conclusion is consistent with previous studies showing that microbial community structure shifts according to the N fertiliser applied (Fierer et al. 2012; Zhou et al. 2015). To further evaluate the major environmental factors shaping the microbial community structure in our soil samples, an RDA was performed based on all of the detected OTUs and the measured soil properties. The results of this analysis indicate that the microbial community composition and structure of our sweet maize/soybean strip intercropping system with two different fertiliser rates was closely correlated with selected variables, namely pH, NH4+, available P, and total K. Taken together, it appears that differences in the chemical properties of the soil can be caused by many factors, such as N fertiliser application and crop management, and subsequently shape the soil microbial community and structure in the root-rhizosphere ecosystem (Ribbons et al. 2016).
In addition to the effects of strip intercropping, N fertilization rate, and duration on the general characteristics of the microbial community, we also analysed the changes in the functional soil microbial groups involved in processes related to N-cycling. We observed that the copy numbers of six key functional genes involved in N-cycling were altered over the course of this 3-year experiment. This result is in accordance to previous studies showing that N-cycling genes were changed by N application (Kong et al. 2010; Tatti et al. 2014). These data further infer that the cropping patterns and N fertilization rates altered the environmental conditions, which subsequently influenced the abundance of functional N-cycling genes. Notably, the amoA gene was largely unchanged over the 3-year planting and fertilization period. This is consistent with previous research indicating that amoA genes are stable and is not as sensitive to environmental influence as other genes (Prosser and Nicol 2012).
The functionally redundant groups involved in nitrite reduction were analysed using the expression of nirS, nirK, and nosZ, which had a similar response patterns to all of the treatments. This further confirmed that N fertilization promotes denitrification in agriculture soils (Xu et al. 2014; Coskun et al. 2017) and increases the denitrifier population. Notably, the abundance of nirS was greater in the reduced fertilization treatment than it was in the conventional fertilization treatment. This phenomenon can largely be explained by the restriction of denitrification in agriculture soils by N application (Kong et al. 2010). The present study also showed that the abundance of the nirK groups significantly increased with N fertiliser, while nirK groups appear to be more related with TN, indicating distinct differences between nirK and nirS denitrifiers in the rhizosphere (Philippot et al. 2007).
In addition, nifH expression was also significantly influenced by cropping pattern and N fertilization rate, indicating that the number of nitrogen-fixing microorganisms increased in the sweet maize/soybean intercropping system. These data suggest that intercropping with reduced N fertilization might increase biological nitrogen fixation. nifH gene abundance has been positively correlated with cropping system and nitrogen rates, it was a consequently result of a combined influence by strip intercropping with soybean and reducing N fertilizer application over time. Moreover, chiA expression was increased in the reduced N application treatments compared to that in the conventional application treatments, as decomposition of chitin contributes to both the labile carbon and nitrogen pools in the soil, it indicating that the potential of N transformation from organic nitrogen into inorganic nitrogen (more available by plants) was higher in the reduced nitrogen application (Lindsay et al. 2010). However, the abundance of nifH gene increased significantly do not mean they are always functionally involved in soil nitrogen fixation. Although we do not determine all of functional N-cycling genes in this study, the six functional genes represent the major processes of N-cycling in strip intercropping system and provide an insight into factors that relate to bacterial community turnover in the nitrogen cycle under different cropping management practices. In this case, the abundance of N fixer increased while that is negated with higher N fertilizer. This is an interesting response that shows a potential plant-soil-microbial interaction that regulates rates of biological N fixation based on system N demand. In cereal-legume strip intercropping system, increased N fertilization is usually benefit to the cereal through their efficiency complementary usage of the available N in the soil. Whereas, the legume also be affected by high N input in strip intercropping system because the biological N fixation may inhibit by the higher mineral N available in the soil. In accordance with many previous studies, which showing that the amount of the biological N fixation is higher in low N fertilizer addition than in high N fertilizer addition systems (Li et al. 2003; Corre-Hellou et al. 2007; Chu et al. 2004).
Conclusions
In this study, we utilized high-throughput sequencing to reveal the effects of N fertiliser reduction and strip intercropping patterns on the bacterial community structure of the soil and on gene expression related to N transformation in sweet maize fields. Although microbial diversity in the sweet maize rhizosphere was relatively stable over 3-year experiment, the composition and structure of the microbial community were markedly modified over time. The expression of six functional genes involved in N transformation increased significantly over the 3-year experimental period, and this increased expression might explain the lower N2O emissions and lower mineral N loss in this study, the previous study conducted in the same field by Tang et al. (2017) showed that this intercrop system could lower soil mineral nitrogen and yield-scaled soil N2O emissions, as the dynamic interactions between roots and rhizosphere might have stimulated the N-involved microbial groups and promoted N transformation processes in the field (Li et al. 2016; Brooker et al. 2015). Therefore, the soil microbial community might benefit from reduced N application via enhanced biological nitrogen fixation in the sweet maize field. Accordingly, legume-cereal strip intercropping may sustain the yield of both crops under low N inputs (Hauggaard-Nielsen et al. 2008) and further improve benefits for agro-environmental systems (Martin-Guay et al. 2018).
References
Bainard LD, Koch AM, Gordon AM, Klironomos JN (2013) Growth response of crops to soil microbial communities from conventional monocropping and tree-based intercropping systems. Plant Soil 363:345–356
Bao SD (2013) Soil agro-chemistrical analysis, 3rd edn. China Agriculture Press, Beijing
Berthrong ST, Yeager CM, Gallegos-Graves L, Steven B, Eichorst SA, Jackson RB, Kuske CR (2014) Nitrogen fertilization has a stronger effect on soil nitrogen-fixing bacterial communities than elevated atmospheric CO2. Appl Environ Microb 80:3103–3112
Brooker RW, Bennett AE, Cong W, Daniell TJ, George TS, Hallett PD, Hawes C, Iannetta PPM, Jones HG, Karley AJ, Li L, McKenzie BM, Pakeman RJ, Paterson E, Schöb C, Shen J, Squire G, Watson CA, Zhang C, Zhang F, Zhang J, White PJ (2015) Improving intercropping: a synthesis of research in agronomy, plant physiology and ecology. New Phytol 206(1):107–117
Campbell BJ, Poison SW, Hanson TE, Mack MC, Schuur EAG (2010) The effect of nutrient deposition on bacterial communities in Arctic tundra soil. Environ Microb 12:1842–1854
Chu GX, Shen Q, Cao J (2004) Nitrogen fixation and N transfer from peanut to rice cultivated in aerobic soil in an intercropping system and its effect on soil N fertility. Plant Soil 263:17–27
Cong WF, Hoffland E, Li L, Six J, Sun JH, Bao XG, Zhang FS, Van Der Werf W (2015) Intercropping enhances soil carbon and nitrogen. Glob Change Biol 21:1715–1726
Correa-Galeote D, Bedmar EJ, Fernandezlopez M, Arone GJ (2016) Bacterial communities in the rhizosphere of amilaceous maize (Zea mays L.) as assessed by pyrosequencing. Front Plant Sci 7(e197):1016
Corre-Hellou G, Brisson N, Launay M, Fusteca J, Crozata Y (2007) Effect of root depth penetration on soil nitrogen competitive interactions and dry matter production in pea–barley intercrops given different soil nitrogen supplies. Field Crops Res 103:76–85
Coskun D, Britto DT, Weiming S, Kronzucker HJ (2017) Nitrogen transformations in modern agriculture and the role of biological nitrification inhibition. Nat Plants 3:17074
Duchene O, Vian J-F, Celette F (2017) Intercropping with legume for agroecological cropping systems: complementarity and facilitation processes and the importance of soil microorganisms—a review. Agr Ecosyst Environ 240:148–161
Fadrosh DW, Ma B, Gajer P, Sengamalay N, Ott S, Brotman RM, Ravel J (2014) An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome 2:6
Fan FL, Li ZJ, Steven AW, Wakelin Y, Liang YC (2012) Mineral fertilizer alters cellulolytic community structure and suppresses soil cellobiohydrolase activity in a long-term fertilization experiment. Soil Biol Biochem 55:70–77
Fierer N, Lauber CL, Ramirez KS, Zaneveld J, Bradford MA, Knight R (2012) Comparative metagenomic, phylogenetic and physiological analyses of soil microbial communities across nitrogen gradients. ISME J 6:1007–1017
Gaba S, Lescourret F, Boudsocq S, Enjalbert J, Hinsinger P, Journet E-P, Navas M-L, Wery J, Louarn G, Malézieux E, Pelzer E, Prudent M, Ozier-Lafontaine H (2015) Multiple cropping systems as drivers for providing multiple ecosystem services: from concepts to design. Agron Sustain Dev 35:607–623
Hai B, Diallo NH, Sall S, Haesler F, Schauss K, Bonzi M, Assigbetse K, Chotte J-L, Munch JC, Schloter M (2009) Quantification of key genes steering the microbial nitrogen cycle in the rhizosphere of sorghum cultivars in tropical agroecosystems. Appl Environ Microb 75:4993–5000
Hargreaves SK, Williams RJ, Hofmockel KS (2015) Environmental filtering of microbial communities in agricultural soil shifts with crop growth. PLoS ONE 10(7):e0134345
Hauggaard-Nielsen H, Jørnsgaard B, Kinane J, Jensen ES (2008) Grain legume–cereal intercropping: the practical application of diversity, competition and facilitation in arable and organic cropping systems. Renew Agric Food Syst 23:3–12
Hink L, Gubry-Rangin C, Nicol GW, Prosser JI (2018) The consequences of niche and physiological differentiation of archaeal and bacterial ammonia oxidisers for nitrous oxide emissions. ISME J 12:1084–1093
Kong AYY, Hristova K, Scow KM, Six J (2010) Impacts of different N management regimes on nitrifier and denitrifier communities and N cycling in soil microenvironments. Soil Biol Biochem 42(9):1523–1533
Lauber CL, Strickland MS, Bradford MA, Fierer N (2008) The influence of soil properties on the structure of bacterial and fungal communities across land-use types. Soil Biol Biochem 40:2407–2415
Li L, Zhang FS, Li XL, Christie P, Sun JH, Yang SC, Tang CX (2003) Interspecific facilitation of nutrient uptake by intercropped maize and faba bean. Nutr Cycl Agroecosyst 65:61–71
Li L, Tilman D, Lambers H, Zhang FS (2014) Plant diversity and overyielding: insights from belowground facilitation of intercropping in agriculture. New Phytol 203:63–69
Li B, Li YY, Wu HM, Zhang FF, Li CJ, Li XX, Lambers H, Li L (2016) Root exudates drive interspecific facilitation by enhancing nodulation and N2 fixation. Proc Natl Acad Sci 113(23):6496–6501
Lindsay EA, Colloff MJ, Gibb NL, Wakelin SA (2010) The abundance of microbial functional genes in grassy woodlands is influenced more by soil nutrient enrichment than by recent weed invasion or livestock exclusion. Appl Environ Microb 76:5547–5555
Martin-Guay M-O, Paquette A, Dupras J, Rivest D (2018) The new green revolution: sustainable intensification of agriculture by intercropping. Sci Total Environ 615:767–772
Philippot L, Hallin S, Schloterm M (2007) Ecology of denitrifying prokaryotes in agricultural soil. Adv Agron 96:135–190
Philippot L, Spor A, Henault C, Bru D, Bizouard F, Jones CM (2013) Loss in microbial diversity affects nitrogen cycling in soil. ISME J 7:1609–1619
Prosser JI, Nicol GW (2012) Archaeal and bacterial ammonia-oxidisers in soil: the quest for niche specialisation and differentiation. Trends Microbiol 20(11):523–531
Ribbons RR, Levy-Booth DJ, Masse J, Grayston SJ, McDonald MA, Vesterdal L, Prescott CE (2016) Linking microbial communities, functional genes and nitrogen-cycling processes in forest floors under four tree species. Soil Biol Biochem 103:181–191
Shen JP, Zhang LM, Guo JF, Jessica LR, He Z (2010) Impact of long-term fertilization practices on the abundance and composition of soil bacterial communities in Northeast China. Appl Soil Ecol 46:119–124
Shi P, Wang SP, Jia S, Gao Q (2015) Effect of 25-year fertilization on soil microbial biomass and community structure in a continuous maize cropping system. Arch Agron Soil Sci 61:1303–1317
Tang YL, Yu LL, Guan AM, Zhou XY, Wang ZG, Guo YG, Wang JW (2017) Soil mineral nitrogen residual and yield-scaled soil N2O emissions lowered by reducing nitrogen application and intercropping with soybean for sweet maize production in southern China. J Integr Agr 16(11):2586–2596
Tatti E, Goyer C, Chantigny M, Wertz S, Zebarth BJ, Burton DL, Filion M (2014) Influences of over winter conditions on denitrification and nitrous oxide-producing microorganism abundance and structure in an agricultural soil amended with different nitrogen sources. Agr Ecosyst Environ 183:47–59
Wallenstein MD, Myrold DD, Firestone M, Voytek M (2006) Environmental controls on denitrifying communities and denitrification rates: insights from molecular methods. Ecol Appl 16(6):2143–2152
Williamson N, Brian P, Wellington EMH (2000) Molecular detection of bacterial and streptomycete chitinases in the environment. Anton Leeuw Int J 78(3):315–321
Xu M, Zhang Q, Xia C, Zhong Y, Sun G, Guo J, Yuan T, Zhou J, He Z (2014) Elevated nitrate enriches microbial functional genes for potential bioremediation of complexly contaminated sediments. ISME J 8:1932–1944
Xue K, Wu L, Deng Y, He Z, Van Nostrand J, Robertson PG, Schmidt TM, Zhou J (2013) Functional gene differences in soil microbial communities from conventional, low-input, and organic farmlands. Appl Environ Microb 79:1284–1292
Zhou J, Guan D, Zhou B, Zhao B, Maa M, Qin J, Jiang X, Chen S, Cao F, Shen D, Li J (2015) Influences of 34-years of fertilization on bacterial communities in an intensively cultivated black soil in northeast China. Soil Biol Biochem 90:42–51
Zhu J, van der Werf W, Anten NPR, Vos J, Evers JB (2015) The contribution of phenotypic plasticity to complementary light capture in plant mixtures. New Phytol 207:1213–1222
Acknowledgements
A sincere thank you to Bernd Wollenweber for his diligent proofreading of this manuscript. And also we would like to thank Chen Xingyuan, Chen Peishou and Xie Zhengsheng for helping us to manage sweet maize cultivation in the field. We are grateful to the constructive comments from editor and two anonymous reviewers on this manuscript.
Funding
This work was supported by the National Natural Science Foundation of China [Grant Nos. 31770556, 31600348]; the Key Technologies R&D Program of China during the 12th five-year Plan period [Grant Nos. 2012BAD14B16-04]; and the Science and Technology Development Program of Guangdong [Grant Nos. 2015B090903077].
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yu, L., Tang, Y., Wang, Z. et al. Nitrogen-cycling genes and rhizosphere microbial community with reduced nitrogen application in maize/soybean strip intercropping. Nutr Cycl Agroecosyst 113, 35–49 (2019). https://doi.org/10.1007/s10705-018-9960-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10705-018-9960-4