
Abstract
In addition to the associated cutaneous and pulmonary manifestations, individuals with the Birt-Hogg-Dubé (BHD) syndrome have an increased risk of developing kidney cancer, which is often bilateral and multifocal. The risk of developing a renal tumor in this population does not decrease with age and therefore warrants a lifelong screening approach. We recommend abdominal imaging every 36 months in individuals without renal lesions at initial screening. Once renal tumors are identified, they should be followed with interval imaging studies until the largest tumor reaches 3 cm in maximal diameter, at which point nephron-sparing surgery should be ideally pursued. While the histology of renal tumors can vary in the BHD syndrome, most tumors possess a relatively indolent natural history and do not require adjuvant therapy if resected when localized to the kidney. With this approach, the vast majority of patients will achieve a curative oncologic outcome and avoid the medical sequelae of chronic renal insufficiency that could otherwise result from total nephrectomy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Originally described in 1977, the Birt-Hogg-Dubé (BHD) syndrome is an autosomal dominant genodermatosis manifested by the development of benign cutaneous lesions, particularly fibrofolliculomas (FF) [1]. Affected individuals are also at risk of developing pulmonary cysts with associated spontaneous pneumothoraces, benign renal cysts, renal cell carcinoma (RCC), and other less common findings (lipomas, parathyroid adenomas, parotid gland tumors, and colonic polyps/tumors) [2–5]. At the National Cancer Institute (NCI), we have been evaluating patients affected with BHD for almost two decades and have developed the following algorithm for the diagnosis and management of BHD-associated kidney cancer.
The mutated gene for BHD, FLCN, was localized and subsequently identified by performing genetic linkage analysis in BHD families [6, 7]. This gene is now known to encode the protein folliculin (FLCN), which acts as a tumor suppressor and has been shown to interact with the mTOR and AMPK signaling pathways (Fig. 1) [7, 8]. While not all patients with confirmed BHD syndrome will develop RCC, our experience suggests that the risk does not decrease with advancing age and thus, lifelong screening is recommended. Herein, we will review our clinical approach to these patients with a particular focus on BHD-associated RCC, including imaging characteristics, surgical strategies, histologic features, and outcome trends.

Presentation and management
The majority of patients with a germline FLCN mutation have dermatologic manifestations and 90 % of families with confirmed BHD syndrome were found to have histologically confirmed cutaneous FF [9]. Other dermatologic lesions that are commonly associated with BHD syndrome include angiofibromas, trichodiscomas, and perifollicular fibromas. The presence of FF should prompt genetic testing to establish the diagnosis. Of note, a minority of BHD patients will not have the typical cutaneous manifestations, but continue to carry the risk of development of RCC and pneumothoraces. At least one BHD kindred in our patient cohort without cutaneous manifestations revealed 3 patients with RCC and 8 patients with pulmonary cysts [10]. Clinicians should thus be aware that the presence of chromophobe/oncocytic renal neoplasms, especially if bilateral or multifocal, should prompt evaluation for the presence of BHD syndrome, even if dermatologic lesions are absent. Likewise, BHD should be included on the differential diagnosis of pulmonary cystic disease.
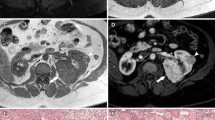
Once the diagnosis of BHD syndrome is established, serial baseline chest and abdominal imaging should be initiated. Computerized tomography (CT) of the chest will identify cysts within the lung or occult pneumothoraces. Abdominal CT or magnetic resonance imaging (MRI) with intravenous contrast provide the best anatomic detail of the kidneys and allow for characterization of any cystic or solid renal lesions (Fig. 2). Ultrasonography has been suggested as a modality for screening or surveillance of BHD-associated renal lesions. Our experience suggests that ultrasonography can often miss BHD-associated lesions, likely due to the similar echogenicity of hybrid oncocytic and chromophobe tumors to the surrounding renal parenchyma. Because of these findings, we do not routinely use renal ultrasound for surveillance of BHD-associated renal lesions and we do not recommend it as a screening modality for the routine detection of renal tumors in patients affected with BHD. In our experience, approximately one-third of BHD patients we have screened were found to have renal tumors on initial abdominal imaging [9]. For our patients without renal lesions on initial imaging, we recommend renal imaging every 36 months for surveillance.

Several examples of diagnostic abdominal imaging demonstrating BHD-associated renal masses. a CT abdomen with intravenous contrast demonstrating a 2.8 cm solid lesion in the lateral mid-pole of the left kidney. b MRI abdomen with intravenous contrast demonstrating a 6.3 cm solid lesion in the anterior aspect of the left kidney. c CT abdomen with intravenous contrast depicting bilateral multifocal renal masses, the largest measuring 6.5 cm in largest diameter
Those individuals who are found to have renal lesions are closely followed as long as the dominant lesion is less than 3 cm in diameter. While unifocal lesions are not uncommon, renal lesions in BHD patients tend to be bilateral and multifocal (Fig. 2c). In our series published in 2005, 27.4 % of the screened individuals were identified to have renal tumors, of which 65 % had multifocal disease. Mixed lesions with both solid and cystic components can be seen. The longest diameter of each renal lesion, as well as an estimation of the percentage of the solid component in a mixed lesion, should be determined.
Our approach for management depends on the lesion size, location, growth rate, number of tumors, and the patient’s general condition (i.e. age, comorbidities, previous surgeries, etc.). In general, surgical resection is recommended once a solid lesion (or solid portion in a mixed lesion) exceeds the 3 cm threshold. Each kidney is considered individually; surgical management is recommended only on the kidney with a dominant tumor exceeding the 3 cm diameter threshold. Although the choice of surgical approach is dependent on the experience and preference of the operating surgeon, in patients with bilateral tumors greater than 3 cm, we most often perform staged surgical procedures in two separate settings. If radical nephrectomy is required on one side and partial nephrectomy on the other side, we most often recommend performing the partial nephrectomy first. Once this kidney has functionally recovered from the partial nephrectomy and the patient has appropriately convalesced, we recommend proceeding with radical nephrectomy on the contralateral unit.
Preoperative pulmonary assessment is recommended and excessive positive pressure ventilation should be avoided intraoperatively to avoid rupture of a pulmonary cyst, which could result in an associated tension pneumothorax. Nephron-sparing surgery, which offers the best chance at renal preservation without comprising oncologic outcomes, is most often recommended (Fig. 3) [11]. Partial nephrectomy can be performed through either an open or laparoscopic approach, the latter of which can be performed with or without the assistance of a surgical robot. Minimally invasive partial nephrectomy has been shown to result in decreased intraoperative blood loss and robotic assistance can be helpful during the renorrhaphy because of the increased dexterity afforded by the robotic arms compared to pure laparoscopy [12]. The choice of surgical approach is based on preoperative assessment of surgical complexity and the surgeon’s experience, without compromising the tenets of an oncological operation. A minimally invasive approach should not be prioritized over cancer control. In addition, referral to a center of excellence for partial nephrectomy is preferable to total nephrectomy in these patients given their hereditary propensity for developing numerous tumors in a lifetime.

44 year-old male with BHD and a history of bilateral renal masses, for which he underwent staged bilateral partial nephrectomy in 2011. a Preoperative CT of the abdomen demonstrating a 7.5 cm mass (red arrow) and a 3.2 cm mass (green arrow) in the right kidney, as well as several smaller masses elsewhere (not shown). b CT of the abdomen after right partial nephrectomy showing successful resection of both previously-identified masses
Once surgery is recommended, our surgical procedure involves removal of all detectable lesions; patients affected with BHD are at risk for the development of many tumors in each kidney. While the shortcomings of ex vivo ultrasonography for the clinical identification and surveillance of BHD renal lesions have been mentioned, we do recommend the utilization of real-time intraoperative renal ultrasound during partial nephrectomy to help detect small endophytic tumors that would otherwise be unidentifiable by gross visual inspection or palpation [10]. Ipsilateral lymph node dissection is not routinely performed, unless lymphadenopathy is detected by pre-operative imaging or intraoperative inspection. Regional lymphadenctomy may also be performed if the primary tumor is large (>4 cm) or preoperative biopsy (if performed) suggests a high-grade clear cell RCC. Most often, BHD renal tumors are enucleated or resected with a very small margin of normal tissue in order to preserve the maximum volume of unaffected renal parenchyma and future renal function. However, if a high-grade clear cell RCC is detected, a wider surgical margin may be advisable.
We do not generally recommend ablative therapies, such as cryotherapy or radiofrequency ablation (RFA), to healthy patients with BHD-associated renal tumors who are appropriate candidates for surgery. While these modalities may be suitable for patients with unifocal renal lesions, particularly if medically unfit for surgery, BHD patients often have multifocal lesions that often require an extensive surgical procedure to remove all of the detectable kidney tumors. Moreover, in patients who have new tumor development or recurrence in the future, previous ablation can significantly complicate subsequent surgical endeavors and lead to higher rates of perioperative complications and total nephrectomy [13, 14]. Furthermore, it is well documented that post-ablation imaging can be challenging to interpret and imaging characteristics in this setting have not been well validated at this time [15]. Consequently, since BHD patients are at lifelong risk for the development of new tumors, our experience suggests that previous RFA or cryotherapy can complicate both the long term evaluation and if needed, subsequent surgical management of this patient population. Nevertheless, cryotherapy or RFA may be an option for BHD patients who are elderly or who are not surgical candidates because of other morbidities.
Gross and microscopic pathology
In BHD patients who underwent surgery at the NCI, a median of 5 tumors were removed (range 1–22 tumors) [10]. The histology of renal tumors from patients with BHD is quite heterogeneous. The largest histologic subtype is the so-called “hybrid oncocytic tumor,” which demonstrates elements of both oncocytoma and chromophobe RCC (Fig. 4). Other histologies (listed in decreasing order of incidence) including chromophobe RCC, clear-cell RCC, oncocytoma, and papillary RCC can also be seen. In the adjacent renal parenchyma, scattered areas of renal oncocytosis (poorly circumscribed lesions consisting of abnormal cells with eosinophilic cytoplasm and large nuclei with stippled heterochromatin) are often identified [16]. These findings suggests that the entire renal parenchyma is at risk for the development of a future renal tumor.

Histologic examples of a BHD-associated hybrid oncocytic tumor. a Low power view of a hybrid nodule showing good circumscription and demarcation from the adjacent normal parenchyma (hematoxylin and eosin stain, ×100). b High power view of oncocytic cells with eosinophilic cytoplasm and clear cells (hematoxylin and eosin stain, ×200)
Clear-cell RCC tends to exhibit a more aggressive phenotype than the other renal tumor histologies seen in the BHD syndrome. Molecular analysis of clear-cell tumors from BHD-affected individuals demonstrated loss of the short arm of chromosome 3 and mutations in the VHL gene in the second allele, which is the classic genetic change seen in clear-cell RCC [16]. How mutations in the BHD gene confer an increased risk to the development clear-cell RCC remains under investigation.
Outcomes
The majority of patients with BHD-associated RCC have an excellent prognosis if managed with nephron-sparing surgery once the dominant tumor reaches 3 cm. With this approach, most individuals will only require a single intervention on a renal unit during their lifetime, which is consistent with the overall indolent nature of BHD-associated RCC. In our experience, we have had only a few patients who have progressed to metastatic disease. Two of these patients were found to have clear-cell RCC on pathology, which tends to be more aggressive than the more common hybrid-oncoctyic and chromophobe histologies in this patient population. Moreover, the tumors were both locally advanced (both 8 cm in diameter) at initial presentation, which is beyond the recommended size for surgical removal [10].
A critical clinical issue in these individuals is the morbidity associated with surgery, particularly the risk of chronic renal insufficiency. The vast majority of BHD patients can be managed with nephron-sparing surgery, which should have little detriment to overall renal function. Clinicians who are not comfortable performing partial nephrectomy on patients with such complicated tumor anatomy may consider referral to a tertiary institution with experience in complex partial nephrectomy. Radical nephrectomy should be reserved only for cases where partial nephrectomy would result in an inferior oncologic outcome or a non-functioning kidney remnant.
Screening recommendations and conclusions
Patients affected with Birt-Hogg-Dubé are at risk for the development of FF, pulmonary cysts, and kidney tumors. Patients suspected of being affected with BHD, such as those with FF, pulmonary cysts or certain types of renal tumors (most notably, hybrid-oncocytic RCC) are recommended to undergo FLCN germline mutation testing (Table 1). In our experience, FLCN germline mutation testing is positive in over 96 % of BHD families [17, 18]. The youngest patient with BHD in whom we have detected kidney cancer was 24 years of age. As such, we currently recommend genetic testing starting at age 21. At-risk individuals who undergo certain activities that could heighten the risk of spontaneous pneumothoraces (i.e. scuba diving) may want to consider genetic testing at an earlier age. Individuals we evaluate who are affected with or at risk for BHD undergo a dermatologic evaluation and a chest CT and an abdominal MRI or CT examination. BHD patients have a lifelong risk of developing RCC and abdominal imaging is recommended at least every 36 months. We prefer MRI because of the high resolution images obtained without subjecting patients to the cumulative radiation exposure of serial CT imaging. We do not recommend subsequent dermatology consultation unless there is a question about the diagnosis or for cosmetic/symptomatic treatment. After the initial screening, we do not recommend periodic chest imaging (CT or MRI) in our asymptomatic patients without pneumothorax. We also do not recommend routine pulmonary medicine or thoracic surgery consultation unless the patient is symptomatic or has a pneumothorax. Although BHD-associated RCC tends to be indolent, it can progress to metastatic disease if not managed appropriately. The interval for follow-up abdominal imaging in patients found to have renal masses smaller than 3 cm is dependent on the location and growth rate of the tumors and is determined by the urologic surgeon managing the patient. Recommendations for surgical intervention depend on the size and location of the tumor and its associated growth rate; surgical intervention is recommended once the largest tumor reaches 3 cm in diameter. BHD-associated tumors are most often amenable to nephron-sparing surgery, which can help prevent chronic renal insufficiency in this patient population.
References
Birt AR, Hogg GR, Dube WJ (1977) Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol 113:1674–1677
Chung JY, Ramos-Caro FA, Beers B, Ford MJ, Flowers F (1996) Multiple lipomas, angiolipomas, and parathyroid adenomas in a patient with Birt-Hogg-Dube syndrome. Int J Dermatol 35:365–367
Roth JS, Rabinowitz AD, Benson M, Grossman ME (1993) Bilateral renal cell carcinoma in the Birt-Hogg-Dube syndrome. J Am Acad Dermatol 29:1055–1056
Toro JR, Glenn G, Duray P, Darling T, Weirich G, Zbar B et al (1999) Birt-Hogg-Dube syndrome: a novel marker of kidney neoplasia. Arch Dermatol 135:1195–1202
Zbar B, Alvord WG, Glenn G, Turner M, Pavlovich CP, Schmidt L et al (2002) Risk of renal and colonic neoplasms and spontaneous pneumothorax in the Birt-Hogg-Dube syndrome. Cancer Epidemiol Biomarkers Prev 11:393–400
Schmidt LS, Warren MB, Nickerson ML, Weirich G, Matrosova V, Toro JR et al (2001) Birt-Hogg-Dube syndrome, a genodermatosis associated with spontaneous pneumothorax and kidney neoplasia, maps to chromosome 17p11.2. Am J Hum Genet 69:876–882
Nickerson ML, Warren MB, Toro JR, Matrosova V, Glenn GM, Turner ML et al (2002) Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dube syndrome. Cancer Cell 2:157–164
Vocke CD, Yang Y, Pavlovich CP, Schmidt LS, Nickerson ML, Torres-Cabala CA et al (2005) High frequency of somatic frameshift BHD gene mutations in Birt-Hogg-Dube-associated renal tumors. J Natl Cancer Inst 97:931–935
Toro JR, Wei MH, Glenn GM, Weinreich M, Toure O, Vocke C et al (2008) BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dube syndrome: a new series of 50 families and a review of published reports. J Med Genet 45:321–331
Pavlovich CP, Grubb RL III, Hurley K, Glenn GM, Toro J, Schmidt LS et al (2005) Evaluation and management of renal tumors in the Birt-Hogg-Dube syndrome. J Urol 173:1482–1486
Shuch B, Singer EA, Bratslavsky G (2012) The surgical approach to multifocal renal cancers: hereditary syndromes, ipsilateral multifocality, and bilateral tumors. Urol Clin North Am 39:133–148, v
Gill IS, Kavoussi LR, Lane BR, Blute ML, Babineau D, Colombo JR Jr et al (2007) Comparison of 1,800 laparoscopic and open partial nephrectomies for single renal tumors. J Urol 178:41–46
Kowalczyk KJ, Hooper HB, Linehan WM, Pinto PA, Wood BJ, Bratslavsky G (2009) Partial nephrectomy after previous radio frequency ablation: the national cancer institute experience. J Urol 182:2158–2163
Nguyen CT, Campbell SC (2008) Salvage of local recurrence after primary thermal ablation for small renal masses. Expert Rev Anticancer Ther 8:1899–1905
Wile GE, Levendecker JR, Krenbiel KA, Dyer RB, Zagoria RJ (2013) CT and MR imaging after imaging-guided thermal ablation of renal neoplasms. Radiographics 27:325–329
Pavlovich CP, Walther MM, Eyler RA, Hewitt SM, Zbar B, Linehan WM et al (2002) Renal tumors in the Birt-Hogg-Dube syndrome. Am J Surg Pathol 26:1542–1552
Schmidt LS, Nickerson ML, Warren MB, Glenn GM, Toro JR, Merino MJ et al (2005) Germline BHD-mutation spectrum and phenotype analysis of a large cohort of families with Birt-Hogg-Dubé syndrome. Am J Hum Genet 76:1023–1033
Benhammou JN, Vocke CD, Santani A, Schmidt LS, Baba M, Seyama K et al (2011) Identification of intragenic deletions and duplication in the FLCN gene in Birt-Hogg-Dube syndrome. Genes Chromosomes Cancer 50:466–477
Baba M, Hong SB, Sharma N, Warren MB, Nickerson ML, Iwamatsu A et al (2006) Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc Natl Acad Sci USA 103:15552–15557
Hasumi H, Baba M, Hong SB, Hasumi Y, Huang Y, Yao M et al (2008) Identification and characterization of a novel folliculin-interacting protein FNIP2. Gene 415:60–67
Hasumi Y, Baba M, Ajima R, Hasumi H, Valera VA, Klein ME et al (2009) Homozygous loss of BHD causes early embryonic lethality and kidney tumor development with activation of mTORC1 and mTORC2. Proc Natl Acad Sci USA 106:18722–18777
Linehan WM, Bratslavsky G, Pinto PA, Schmidt LS, Neckers L, Bottaro DP et al (2010) Molecular diagnosis and therapy of kidney cancer. Ann Rev Med 61:329–343
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. The authors wish to thank Georgia Shaw (editorial and graphics support), Dr. Maria J. Merino (for providing the BHD kidney cancer histology images), and Rabindra Gautam (for providing the radiologic images).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Stamatakis, L., Metwalli, A.R., Middelton, L.A. et al. Diagnosis and management of BHD-associated kidney cancer. Familial Cancer 12, 397–402 (2013). https://doi.org/10.1007/s10689-013-9657-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10689-013-9657-4