
Abstract
Nevoid basal cell carcinoma syndrome (NBCCS) is an autosomal dominant disorder characterized by developmental defects and tumorigenesis. The gene responsible for NBCCS is PTCH1, encoding a receptor for the secreted protein, sonic hedgehog. Recently, a Chinese family with NBCCS carrying a missense mutation in PTCH2, a close homolog of PTCH1, was reported. However, the pathological significance of missense mutations should be discussed cautiously. Here, we report a 13-year-old girl diagnosed with NBCCS based on multiple keratocystic odontogenic tumors and rib anomalies carrying a frameshift mutation in the PTCH2 gene (c.1172_1173delCT). Considering the deleterious nature of the frameshift mutation, our study further confirmed a causative role for the PTCH2 mutation in NBCCS. The absence of typical phenotypes in this case such as palmar/plantar pits, macrocephaly, falx calcification, hypertelorism and coarse face, together with previously reported cases, suggested that individuals with NBCCS carrying a PTCH2 mutation may have a milder phenotype than those with a PTCH1 mutation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Nevoid basal cell carcinoma syndrome (NBCCS) (OMIM 109400), also known as Gorlin syndrome, is an autosomal dominant disorder characterized by developmental defects including bifid ribs, palmar or plantar pits, and tumorigenesis such as the development of basal cell carcinoma, medulloblastoma, or keratocystic odontogenic tumor (KCOT) (formerly known as odontogenic keratocysts) [1]. It is transmitted with complete penetrance and variable expressivity. The gene responsible for NBCCS is the human homologue of the Drosophila patched gene, PTCH1 [2, 3]. The human PTCH1 gene contains 23 coding exons spanning approximately 70 kb and encodes a protein of 1,447 amino-acid residues containing 12 transmembrane-spanning domains and two large extracellular loops [2]. The PTCH1 protein is the ligand-binding component of the sonic hedgehog (Shh) receptor complex. In the absence of Shh binding, PTCH1 is thought to hold smoothened (SMO), a 7-pass transmembrane protein, in an inactive state and thus inhibit signaling to downstream genes. Upon the binding of Shh, the inhibition of SMO is released and signaling is transduced leading to the activation of target genes by the Gli family of transcription factors [4]. Therefore, aberrant activation of the Shh signaling cascade due to the haploinsufficiency of PTCH1 is believed to cause NBCCS.
In vertebrates, there exists a close homolog of PTCH1 named PTCH2. The human PTCH2 gene contains 22 coding exons spanning approximately 15 kb and encodes a protein of 1,203 amino-acid residues [5]. Recently, in six affected members of a Chinese Han family with NBCCS, Fan et al. identified a heterozygous germline missense mutation in the PTCH2 gene [6]. Here we report a case with NBCCS carrying a frameshift mutation due to a 2-bp deletion in PTCH2. To our knowledge, this is the first report of NBCCS caused by a frameshift mutation and the second report of a germline mutation in the PTCH2 gene.
Clinical report
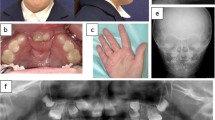
A 13-year-old Japanese girl was referred to our hospital with jaw cysts and a rib abnormality. She was born weighing 2,570 g, with a height of 48 cm and a head circumference of 32 cm. Her two siblings as well as her parents exhibited no similar features. No consanguineous marriage was noticed in her pedigree. At 10 years of age, she had multiple KCOTs (Fig. 1a) and underwent a surgical operation to remove them. At 12 years of age, she exhibited proteinuria, and was diagnosed as having chronic glomerular nephritis. Then, she was referred to our hospital for further investigation. At examination, she was 150.1 cm tall (mean) and weighed 57.8 kg (+1.8SD). Her head circumference was 54.6 cm (mean). She had normal intelligence without neurological deficit. She exhibited no structural abnormalities of face, oral cavity, or limbs. However, chest roentgenogram revealed a left bifid rib without any other bone abnormalities (Fig. 1b). She did not exhibit palmar or plantar pits, falx calcification, medulloblastomas, or basal cell carcinomas at that time. Since she exhibited KCOTs and a rib anomaly fulfilling the diagnostic criteria made by Kimonis et al. (two major criteria), we diagnosed her as having NBCCS.

Roentgenograms of the patient. a Pantomography shows bilateral keratocystic odontogenic tumors (Arrows). b Chest roentgenogram reveals a bifid anomaly of the left sixth rib (Arrow)
Methods
DNA extraction and PCR-sequencing analysis
All experiments described below were approved by the ethics committee at Kitasato University. DNA was extracted from peripheral blood lymphocytes using a QIAamp DNA blood midi kit (QIAGEN). The complete coding region of the PTCH1, PTCH2, suppressor of fused (SUFU) and SMO genes, including all splice junctions, was amplified from constitutional DNA as described previously [7]. Primers used for amplifying PTCH1, PTCH2 and SUFU exons were described previously [5, 7, 8]. Those used for amplifying SMO are listed in supporting information Table 1. Amplified products were gel-purified using a QIAEX II gel extraction kit (QIAGEN) and cycle sequenced with a BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems) in both directions. The sequence was analyzed on a 3130 Genetic Analyzer (Applied Biosystems).
Comparison of clinical manifestations
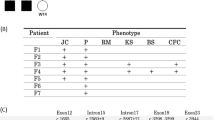
Details of a nationwide survey of NBCCS performed in Japan have been described previously [9]. The survey covered 157 NBCCS patients whose clinical details were available. Major and minor criteria for NBCCS proposed by Kimonis et al. [10] were evaluated in these patients and compared with those observed in patients carrying a PTCH2 mutation reported previously including the present case [6].
Results
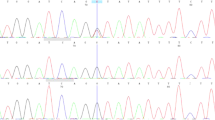
No mutation in PTCH1 or the related genes SUFU and SMO was detected in the peripheral blood from this patient. Although deletion of the entire PTCH1 gene is a common event in point mutation-negative cases of NBCCS as we reported previously [11], no such deletion was observed using either a ligation-dependent probe amplification method or high-resolution array-based comparative genomic hybridization technology [11–13]. We then sequenced all exons of the PTCH2 gene, since PTCH2 is a close homolog of PTCH1 and is also a suppressor component of the Shh pathway. As a result, a heterozygous 2-base-pair deletion, c.1172_1173delCT, was detected in exon 9 of PTCH2 (Fig. 2). This mutation caused a frameshift and created a premature termination codon (PTC) at the site of the deletion resulting in a truncated form of the PTCH2 protein, p.S391X.

Electropherograms of the PTCH2 exon 9 sequence. DNA extracted from the peripheral blood of the patient (Case) as well as a healthy control (Control) was subjected to PCR direct sequencing. The predicted translation is indicated at the bottom of each electropherogram. PTC created by the deletion is indicated by an asterisk
In order to characterize the phenotype of individuals carrying a PTCH2 mutation, we next evaluated the number of positive criteria for NBCCS proposed by Kimonis et al. [10] in two groups; first, NBCCS patients collected by a nationwide survey described previously [9], second, reported individuals carrying a PTCH2 mutation including our case. In spite of the comparable mean ages in two groups (33.1 vs 34.1 years old), positivities of most of the criteria were lower in the PTCH2 mutation-positive group than in the other, indicating that PTCH2 mutations cause a milder phenotype than the classical NBCCS (Table 1). In fact, only 4 out of 7 individuals in the PTCH2 mutation-positive group diagnosed as having NBCCS according to this diagnostic criteria.
Discussion
NBCCS is caused by a mutation in the gene PTCH1, with rare exceptions in which a SUFU mutation has been identified [8, 14, 15]. Recently, a heterozygous missense mutation in the PTCH2 gene, c.2157G>A (p.R719Q), was identified in 6 affected members of a Chinese Han family with NBCCS [6]. Although the pathological significance of missense mutations should be discussed cautiously, this mutation was demonstrated to result in the inactivation of PTCH2’s inhibitory activities at least in vitro. In this paper, we reported a second germline mutation of PTCH2, c.1172_1173delCT, found in a Japanese patient with NBCCS. This mutation created a PTC at the site of the deletion in the mutant allele, resulting in the truncation of the PTCH2 protein. However, since the PTC leads to the degradation of mRNA via a mechanism called nonsense-mediated mRNA decay [16], a haploinsufficiency of PTCH2 is expected to play an important role in this case.
Interestingly, this mutation is also present in the dbSNP database as rs56126236, submitted by the Center for Genome Medicine, Kyoto University Graduate School of Medicine, Japan. However, details such as frequency are unclear. Therefore, we analyzed 63 healthy Japanese individuals (126 alleles) on this mutation, but found none carrying this deletion. Thus, it is unlikely that this is a rare polymorphism at least in a Japanese population.
However, unfortunately, we were unable to get informed consents from family members and, therefore, could not add data regarding this issue. Nonetheless, considering the deleterious nature of the mutation, we believe that the mutation found in this patient is generated de novo.
Homozygous mutant mice, Ptch2 −/−, developed normally, were viable and fertile, and did not display any obvious defects in hair follicle, limb, neural, or testis development [17]. However, with age, homozygous mutant male mice developed skin lesions consisting of alopecia and epidermal hyperplasia, suggesting a role for Ptch2 in adult epidermal homeostasis via Shh signaling. In accordance with the milder phenotype of Ptch2 −/− than Ptch1 −/−, which is embryonic lethal [18], it is not surprising that individuals carrying a PTCH2 mutation also exhibit milder clinical manifestations than those with classical NBCCS. Our patient lacked typical NBCCS phenotypes such as palmar/plantar pits, falx calcification, macrocephaly, hypertelorism and coarse face, the frequencies of which are 60.1, 79.6, 26.5, 68.8, and 27.9 %, respectively, in the Japanese population [9]. In fact, this case does not fulfill the criteria by Evans et al. [19] because a rib anomaly is considered to be a minor criterion. Since the number of cases with a PTCH2 mutation is still limited, the accumulation of such patients is expected to further clarify their characteristic phenotype.
A genotype–phenotype correlation has not been reported in NBCCS patients [20]. However, mutations in the SUFU gene were reported to result in a much higher incidence of medulloblastoma than those in PTCH1 [8, 14, 21]. It is also reported that a large genomic deletion encompassing PTCH1 leads to NBCCS with atypical clinical manifestations, probably due to a deletion of adjacent gene(s) [12]. Therefore, it should be noted that NBCCS cases caused by a mutation of a gene other than PTCH1 have phenotypes different from those of classical NBCCS caused by PTCH1 mutations.
References
Gorlin RJ (1987) Nevoid basal-cell carcinoma syndrome. Medicine (Baltimore) 66:98–113
Johnson RL, Rothman AL, Xie J, Goodrich LV, Bare JW, Bonifas JM, Quinn AG, Myers RM, Cox DR, Epstein EH Jr, Scott MP (1996) Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science 272:1668–1671
Hahn H, Wicking C, Zaphiropoulous PG, Gailani MR, Shanley S, Chidambaram A, Vorechovsky I, Holmberg E, Unden AB, Gillies S, Negus K, Smyth I, Pressman C, Leffell DJ, Gerrard B, Goldstein AM, Dean M, Toftgard R, Chenevix-Trench G, Wainwright B, Bale AE (1996) Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 85:841–851
Ingham PW, McMahon AP (2001) Hedgehog signaling in animal development: paradigms and principles. Genes Dev 15:3059–3087
Smyth I, Narang MA, Evans T, Heimann C, Nakamura Y, Chenevix-Trench G, Pietsch T, Wicking C, Wainwright BJ (1999) Isolation and characterization of human patched 2 (PTCH2), a putative tumour suppressor gene in basal cell carcinoma and medulloblastoma on chromosome 1p32. Hum Mol Genet 8:291–297
Fan Z, Li J, Du J, Zhang H, Shen Y, Wang CY, Wang S (2008) A missense mutation in PTCH2 underlies dominantly inherited NBCCS in a Chinese family. J Med Genet 45:303–308
Fujii K, Kohno Y, Sugita K, Nakamura M, Moroi Y, Urabe K, Furue M, Yamada M, Miyashita T (2003) Mutations in the human homologue of Drosophila patched in Japanese nevoid basal cell carcinoma syndrome patients. Hum Mutat 21:451–452
Kijima C, Miyashita T, Suzuki M, Oka H, Fujii K (2012) Two cases of nevoid basal cell carcinoma syndrome associated with meningioma caused by a PTCH1 or SUFU germline mutation. Fam Cancer 11:565–570
Endo M, Fujii K, Sugita K, Saito K, Kohno Y, Miyashita T (2012) Nationwide survey of nevoid basal cell carcinoma syndrome in Japan revealing the low frequency of basal cell carcinoma. Am J Med Genet A 158A:351–357
Kimonis VE, Goldstein AM, Pastakia B, Yang ML, Kase R, DiGiovanna JJ, Bale AE, Bale SJ (1997) Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am J Med Genet 69:299–308
Nagao K, Fujii K, Saito K, Sugita K, Endo M, Motojima T, Hatsuse H, Miyashita T (2011) Entire PTCH1 deletion is a common event in point mutation-negative cases with nevoid basal cell carcinoma syndrome in Japan. Clin Genet 79:196–198
Fujii K, Ishikawa S, Uchikawa H, Komura D, Shapero MH, Shen F, Hung J, Arai H, Tanaka Y, Sasaki K, Kohno Y, Yamada M, Jones KW, Aburatani H, Miyashita T (2007) High-density oligonucleotide array with sub-kilobase resolution reveals breakpoint information of submicroscopic deletions in nevoid basal cell carcinoma syndrome. Hum Genet 122:459–466
Kosaki R, Nagao K, Kameyama K, Suzuki M, Fujii K, Miyashita T (2012) Heterozygous tandem duplication within the PTCH1 gene results in nevoid basal cell carcinoma syndrome. Am J Med Genet A 158A:1724–1728
Taylor MD, Liu L, Raffel C, Hui CC, Mainprize TG, Zhang X, Agatep R, Chiappa S, Gao L, Lowrance A, Hao A, Goldstein AM, Stavrou T, Scherer SW, Dura WT, Wainwright B, Squire JA, Rutka JT, Hogg D (2002) Mutations in SUFU predispose to medulloblastoma. Nat Genet 31:306–310
Pastorino L, Ghiorzo P, Nasti S, Battistuzzi L, Cusano R, Marzocchi C, Garre ML, Clementi M, Scarra GB (2009) Identification of a SUFU germline mutation in a family with Gorlin syndrome. Am J Med Genet A 149A:1539–1543
Holbrook JA, Neu-Yilik G, Hentze MW, Kulozik AE (2004) Nonsense-mediated decay approaches the clinic. Nat Genet 36:801–808
Nieuwenhuis E, Motoyama J, Barnfield PC, Yoshikawa Y, Zhang X, Mo R, Crackower MA, Hui CC (2006) Mice with a targeted mutation of patched2 are viable but develop alopecia and epidermal hyperplasia. Mol Cell Biol 26:6609–6622
Goodrich LV, Milenkovic L, Higgins KM, Scott MP (1997) Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 277:1109–1113
Evans DG, Ladusans EJ, Rimmer S, Burnell LD, Thakker N, Farndon PA (1993) Complications of the naevoid basal cell carcinoma syndrome: results of a population based study. J Med Genet 30:460–464
Wicking C, Shanley S, Smyth I, Gillies S, Negus K, Graham S, Suthers G, Haites N, Edwards M, Wainwright B, Chenevix-Trench G (1997) Most germ-line mutations in the nevoid basal cell carcinoma syndrome lead to a premature termination of the PATCHED protein, and no genotype–phenotype correlations are evident. Am J Hum Genet 60:21–26
Brugieres L, Pierron G, Chompret A, Paillerets BB, Di Rocco F, Varlet P, Pierre-Kahn A, Caron O, Grill J, Delattre O (2010) Incomplete penetrance of the predisposition to medulloblastoma associated with germ-line SUFU mutations. J Med Genet 47:142–144
Acknowledgments
This research was supported by Science Research Grants for intractable diseases in Japan (H22-intractable diseases-120) from the Ministry of Health, Labour and Welfare, and by a Grant-in-Aid for Scientific Research (20591261) from the Ministry of Education, Culture, Sports, Science and Technology.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Fujii, K., Ohashi, H., Suzuki, M. et al. Frameshift mutation in the PTCH2 gene can cause nevoid basal cell carcinoma syndrome. Familial Cancer 12, 611–614 (2013). https://doi.org/10.1007/s10689-013-9623-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10689-013-9623-1