
Abstract
Anthracnose, caused by Colletotrichum lentis, is one of the most damaging diseases of lentil (L. culinaris) in western Canada. Lens ervoides accession IG 72815 exhibits high levels of resistance to the pathogenic races 0 and 1. The objective of this study was to identify quantitative trait loci (QTL) associated with anthracnose resistance in lentil using a recombinant inbred line (RIL) population from the interspecific cross between IG 72815 and the susceptible cultivar Eston. A total of 168 RILs were genotyped and evaluated for anthracnose race 0 and race 1 reaction in the growth chamber and polyhouse. QTL analysis identified major resistance loci on chromosomes 3 and 7 for both races, accounting for 20.1–31.2% and 8.3–18.4%, respectively, of the total phenotypic variation for anthracnose resistance. Multiple classes of putative defense-related genes are located within both loci. Further characterization of these regions will facilitate the introgression of anthracnose resistance from Lens ervoides into elite lentil cultivars via marker-assisted selection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lentil (Lens culinaris Medik.) is an economically important pulse crop on a global scale that is consumed for its high levels of protein, micronutrients, vitamins, and dietary fiber (Kissinger 2016; Raghuvanshi and Singh 2009). The crop is cultivated in more than 70 countries, and western Canada accounted for 46% of the world’s lentil production from 2013 to 2017 (FAOSTAT 2017). Lentil productivity in western Canada is challenged primarily by four fungal diseases: ascochyta blight (Ascochyta lentis), stemphylium blight (Stemphylium botryosum), aphanomyces root rot (Aphanomyces euteiches), and anthracnose, caused by the fungal ascomycete pathogen Colletotrichum lentis (Damm) (Damm et al. 2014). Anthracnose can cause up to 70% yield loss under high disease pressure (Chongo et al. 1999; Morrall and Pedersen 1990). The first incidence of anthracnose in lentil was reported in the province of Manitoba in 1987 (Morrall 1988) and is now widespread in western Canada.
The pathogen survives as microsclerotia on lentil debris and primarily spreads between fields by wind. The disease management options include 3–4 year crop rotations, foliar fungicide application and host-plant resistance (Buchwaldt et al. 2018). Two pathogenic races of C. lentis were previously identified (Buchwaldt et al. 2004) and re-designated as race 0 and race 1 (Banniza et al. 2018). Race 0 has high virulence, and no effective resistance has been found in L. culinaris accessions; and race 1 is less virulent, against which partial resistance was identified in a number of L. culinaris accessions (Buchwaldt et al. 2004, 2018; Gela et al. 2020).
Current germplasm collections of lentil exhibit low genetic diversity and a narrow genetic base for anthracnose resistance (Buchwaldt et al. 2004; Gela et al. 2020). Genetic diversity can be reintroduced using crop wild relatives (reviewed by Coyne et al. 2020; Dempewolf et al. 2017). These natural genetic reservoirs retain much of the genetic diversity lost during the process of domestication and/or deliberate selection for cultivar development (Tanksley and McCouch 1997). The species of the genus Lens are classified into four gene pools; the primary gene pool (L. culinaris, L. orientalis and L. tomentosus), secondary (L. odemensis and L. lamottei); tertiary (L. ervoides), and quaternary gene pool (L. nigricans) (see Cubero et al. 2009; Wong et al. 2015). All the species have seven chromosomes (2n = 14) and similar karyotypes (Ladizinsky et al. 1984; Van Oss et al. 1997). Lens ervoides exhibit resistance to many lentil diseases, including resistance to both anthracnose races (Tullu et al. 2006), ascochyta blight (Tullu et al. 2010), and stemphylium blight (Podder et al. 2013). High level of resistance to anthracnose race 0 has not been found in other wild relatives (Barilli et al. 2020). Resistance breeding against the highly virulent race 0 of C. lentis is therefore especially dependent on the use of resistant germplasm from L. ervoides. Initial screening of L. ervoides identified two accessions, L-01-827A and IG 72815 with superior resistance to both races (Tullu et al. 2006; Fiala et al. 2009). Embryo rescue was used to create F1 hybrids that were advanced to develop interspecific RIL population, such as LR-26 (L. culinaris Eston × L. ervoides IG 72815) (Tullu et al. 2013).
Of the available management strategies for anthracnose, the use of genetic sources of host resistance may be the best approach to control the disease. Little is known, however, about the genomic regions and molecular markers associated with anthracnose resistance. Therefore, more precise localization of QTL/genes is critical to the development and application of marker assisted breeding and develop strategies for pyramiding anthracnose resistance genes from multiple resistance sources into elite cultivars. Both dominant and recessive sources of genetic resistance to anthracnose race 1 are reported in L. culinaris intraspecific populations (Tullu et al. 2003; Buchwaldt et al. 2013). In L. culinaris × L. ervoides interspecific populations double recessive genetic inheritance of resistance to race 0 and race 1 are reported (Fiala et al. 2009; Tullu et al. 2013). Bhadauria et al. (2017) mapped QTLs conferring resistance to race 0 and race 1 on chromosomes 2, 3, 5, and 7 using an L. ervoides intraspecific population (L-01-827A × IG 72815). However, these QTLs are not yet mapped in interspecific populations.
In the current study we tested the hypothesis that regions of the L. ervoides genome that are associated with anthracnose resistance will continue to confer resistance following hybridization with L. culinaris. The objective was to identify the regions of the L. ervoides genome associated with anthracnose resistance in the LR-26 interspecific RIL population.
Material and methods
Plant materials
Evaluation of genetic resistance to C. lentis was conducted using 168 RILs of the LR-26 interspecific mapping population derived from a cross between L. culinaris Eston × L. ervoides IG 72815 (Tullu et al. 2013). The RILs had been advanced using single seed descent to the F7 generation. Then the F7-derived bulked seed of the RILs were selfed for at least three additional generations. The L. ervoides accession IG 72815 is from Turkey and has resistance to both anthracnose races (Tullu et al. 2006). Lentil cultivar Eston is a small seeded, yellow cotyledon, green seed coat, early maturing line released in Canada in 1980 (Slinkard 1981); it is susceptible to both races of anthracnose.
Inoculation and phenotyping for anthracnose reactions
Colletotrichum lentis isolates CT-30 (race 0) and CT-21 (race 1) (Banniza et al. 2018) were used to inoculate the LR-26 RIL population in two environments: in growth chambers at the University of Saskatchewan (USask) College of Agriculture and Bioresources phytotron facility, Saskatoon, Canada, and in an outdoor polyhouse. Fungal inoculum production and inoculation in the growth chamber was done according to methods described by Gela et al. (2020). In the growth chambers, the parents and LR-26 RILs were evaluated (inoculated with race 0 isolate CT-30 and race 1 isolate CT-21, separately) for resistance to C. lentis; under polyhouse conditions they were evaluated for resistance to race 0 isolate CT-30. In the growth chamber experiments, plants were grown in 38-cell cone trays (26.8 × 53.5 cm) filled with Sun Gro Horticulture Sunshine Mix LA4 (Sun Gro Horticulture, Bellevue, USA) and perlite (Specialty Vermiculite Canada, Winnipeg, MB) in a 3:1 ratio (v/v). The LR-26 RILs and the parents were randomized in a set of trays per replicate. Four-week-old seedlings were inoculated with a spore suspension (5 × 104 spores mL−1) of C. lentis race 0 and 1 at 3 mL per plant using an airbrush. Plants were placed in an incubation chamber (relative humidity 90–100%) for 48 h before being moved to misting benches (see Gela et al. 2020). The disease severity data were collected per individual plant (an experimental unit). The experiment was repeated eight times for each race separately (blocked over time) and analyzed as a randomized complete block design with eight replications. Individual plants were scored for anthracnose severity at 8–10 days post-inoculation (dpi).
The polyhouse experiment was conducted at the Department of Plant Sciences field laboratory at the USask. Four seeds of each genotype, and two seeds of cultivar Eston (susceptible control), were sown in individual 1-gallon pots (15.5 cm diameter) containing a soilless mixture (Sunshine Mix No. 4, Sun Grow Horticulture® Ltd., Vancouver, BC, Canada). The plants were grown under open field conditions for 6 weeks (early flowering stage). Then a polyhouse tunnel cover of translucent thin plastic sheeting was installed by suspending it 1.5 m above the ground over the pots immediately before inoculation. The area under the cover was equipped with a misting irrigation system. Each pot was sprayed with approximately 36 ml (6 ml plant−1) of aqueous spore suspension (5 × 104 spores mL−1) of race 0 isolate CT-30 until runoff using a pressurized knapsack sprayer. The inoculations were performed in the evening to avoid high temperature conditions and to facilitate the germination of spores on the leaves. After inoculation, misting irrigation was applied starting from early morning to evening for 30 s every 15 min to promote disease development. The experiment was conducted in a randomized complete block design (RCBD) with four replications. Disease severity data were collected 14 d after inoculation using a 0 to 10 rating scale with 10% increments. Data were converted to percentage disease severity using the class midpoints for data analysis.
Statistical analysis
Disease scores data were analyzed using SAS v.9.4 (SAS Institute, Cary, USA). Normality and variance homogeneity of the residuals were tested using the Shapiro–Wilk normality test and Levene’s test for homogeneity, respectively. The data did not fit the assumptions of a Gaussian distribution. As a result, a generalized linear mixed model with a binomial distribution function was fitted to the data using PROC GLIMMIX with the LOGIT link function (SAS 9.4). Means of the disease reaction scores were compared for post hoc comparison using Tukey’s Honestly Significant Difference at α = 0.05. The mean disease score data were transformed using log transformation for QTL analysis to reduce the skewness. Spearman’s rank correlation of disease severity between races and test environments was performed using the CORR procedure in SAS.
Genotyping and linkage mapping
Genotyping was done as described in Chen (2018). Briefly, total genomic DNA (gDNA) from bulk leaf tissue of parents and LR-26 RILs was extracted using the DNeasy® 96 Plant Kit (Qiagen, Germany). The LR-26 RILs were genotyped following the two-enzyme (PstI-MspI)-based GBS protocol of Poland et al. (2012) as described in Wong et al. (2015) for lentil. The gDNA quality was checked on a 1% agarose gel, quantified using PicoGreen, normalized to a concentration of 20 ng/μl per RIL, a total of 200 ng of gDNA per RIL was digested with PstI-HF and MspI, and ligated to barcoded adapters using T4 DNA ligase (New England Biolabs, Inc., Ipswich, USA). Individual libraries were then pooled (43-plexed library), bead-cleaned, PCR amplified, and bead-cleaned again. Average size and concentration of pooled libraries were estimated using a DNA2100 chip on an Agilent Bioanalyzer, and libraries were sequenced on an Illumina HiSeq.2000 instrument at the Génome Québec Innovation Centre, McGill University, Montréal Canada.
The GBS reads were processed using an open source GBS pipeline written in Perl and developed by the Pulse Bioinformatics group in the Department of Plant Sciences, USask, Canada (https://knowpulse.usask.ca/software/GBS-Pipeline; Wong et al. 2015). The pipeline demultiplexes raw reads and removes barcode sequences prior to trimming them with the recommended parameters for paired-end reads using Trimmomatic-0.38 (Bolger et al. 2014). The pipeline then aligns the trimmed reads to the reference genome, in this case L. culinaris cultivar CDC Redberry genome v2.0 (Ramsay et al. 2019), using Bowtie2-2.2.9 (Langmead and Salzberg 2012). The parameters for maximum fragment length were set to 11,000, maximum reseed rate was set to 5, and reads were allowed multimap up to three times before using a custom script to filter for the best hit of three. The final step in the pipeline was variant calling of the combined samples using SAMtools-1.9 and BCFtools-1.6 (Li et al. 2009) using the multiallelic calling model, respectively. Overall, a total of 833,041,263 raw reads of the GBS library were processed and resulted in detection of 167,102 raw SNPs that passed all quality controls. These were further filtered using VCFtools (Danecek et al. 2011) to retain SNPs with less than 35% missing allele calls (minimum read depth = 5) and SNPs with minor allele frequency greater than 25%. Linkage analysis was performed using ASMap (Taylor and Butler 2017) with the parameters: segregation ratio = 75:25 and p value = 10−5; and treating heterozygous calls as missing values.
QTL mapping of disease resistance
Multiple QTL mapping (Manichaikul et al. 2009) and composite interval mapping (Zeng 1994) run in R/qtl software (Broman et al. 2003) were used to detect QTL. The Haley-Knott regression (Haley and Knott 1992) was used for both methods to confirm the consistency of the QTL detected due to the non-normal distribution of the phenotype. The regression-based QTL mapping methods are robust against non-Gaussian trait distribution (Rebai 1997). Multiple QTL mapping was completed with the stepwiseqtl function (Broman et al. 2003) and the optimal QTL model was chosen based on the highest penalized LOD score (Manichaikul et al. 2009) after forward and backward selection and elimination modelling. Penalties for model selection and genome-wide significance threshold (α = 0.05) were determined by 1000 permutations with scantwo function for two-dimensional QTL scan. For composite interval mapping (CIM), five markers were selected as cofactors by forward selection to control genomic background effects. Thresholds for declaring QTL were determined by 1000 permutations at a significance of α = 0.05. The percentage of the phenotypic variance explained (PVE) and effects of QTL were obtained by fitting a mixed linear model using the “fitqtl” function. The confidence intervals for each QTL were estimated using the “lodint” function that calculates the 1.5 LOD support intervals. The QTL intervals on the genetic map were compared against their physical positions on the CDC Redberry genome assembly v.2.0 (Ramsay et al. 2019) for the identification candidate genes associated with disease resistance. All reported disease resistance (R-) or defense-related genes in plants were considered for selection of candidate genes.
Results
Reactions of RILs to C. lentis infection
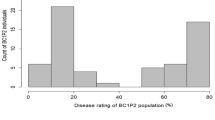
The resistant parent IG 72815 showed moderate to high levels of resistance to both race 0 and race 1 (35% mean disease severity), whereas the susceptible parent Eston showed susceptible reactions to both races (95% mean disease severity) in all assays. Significant variation in disease reaction was observed among the RILs for both races (p < 0.001). A high proportion of the lines in the population showed susceptible reactions to race 0 and race 1 under growth chamber inoculations (Fig. 1a, b). Disease severity scores ranged from 8.33 to 95% and 5 to 95%, with a mean of 79.4% and 76.0% for race 0 and race 1, respectively. The polyhouse ratings of the disease reactions were continuously distributed, with a skew toward the higher level of disease severity, ranging from 12.5 to 95%, with a mean of 65.7% (Fig. 1c). Correlation analysis indicated a significant positive relationship between the results from the two races and test environments (Table 1), suggesting the resistance to both races derived from IG 72815 may be controlled by the same gene or by tightly linked genes. Chi-squared analysis of the combined data suggested that the best-fitting segregation ratio was 1 resistant: 3 susceptible (χ2 = 1.115 at 1 d.f.; p = 0.291), supporting the presence of two complementary resistance genes that confer resistance to anthracnose.

Frequency distributions of percent anthracnose severity for 168 members of the interspecific RIL population LR-26 derived from the interspecific cross Lens culinaris Eston × L. ervoides IG 72815 following inoculation with: a) race 0, and b) race 1 of C. lentis under growth chamber (phytotron) conditions, and c) race 0 in a polyhouse. Disease severity was rated on a 0–10 scale, increasing in 10% increments. Data were converted to % disease severity using the class midpoints for data analysis
Linkage map construction
A genetic map was constructed from 5455 SNP markers that mapped to 7 linkage groups (LGs) (Table 2). The LGs were numbered to match the respective chromosomes where markers lie in the lentil reference genome (v2.0; Ramsay et al. 2019). The linkage map spanned a total genetic distance of 3252.8 cM with an average marker interval at 0.6 cM. The genetic distance within LGs varied from 363.5 cM for LG 3 to 627.4 cM for LG 5. LG 5 contained the smallest number of SNPs, whereas LG 2 and LG 4 contained the highest number of SNPs (Table 2).
QTL for anthracnose resistance
We detected two large effect QTLs associated with anthracnose resistance (Fig. 2); one on chromosome 3, and another on chromosome 7. A genome-wide view of the QTL detected with individual data sets across the chromosomes (LGs) and the details of each QTL identified are presented in Table 3 and Fig. S2. The results were consistent across test conditions and were not sensitive to the QTL analysis methods used. QTL on a chromosome were considered the same QTL when their 1.5-LOD intervals overlapped, although the position of the peaks varied slightly. The QTL on chromosome 3 (qANTH-3) was consistently found in both environments and co-localized in the physical interval of 285.1–322.2 Mb for both races of C. lentis. This QTL explained 20.1 to 31.2% of the phenotypic variation. Similarly, the QTL on chromosome 7 (qANTH-7) was detected in both environments and found to overlap for both races in the interval of 518.7–522.5 Mb and explained 8.3 to 18.4% of the phenotypic variation. As expected, the resistance allele for both qANTH-3 and qANTH-7 were contributed from the wild parent IG 72815, and their attribution to both race 0 and 1 resistance also explained the high correlation observed (r2 = 0.8–0.9) between race 0 and race 1 phenotypes in the LR-26 population (Table 1). The marker effect of tightly linked SNP marker to qANTH-3 (Lcu.2RBY.Chr3_308775097) and qANTH-7 (Lcu.2RBY.Chr7_521279838) were compared with the resistance levels of the LR-26 population (Fig. 3). The homozygous resistant genotype BB (wild parent) is associated with enhanced resistance compared to the homozygous susceptible genotype AA for the combined mean disease scores.

Location of anthracnose resistance QTL in IG 72815 on linkage groups (LG) 3 and 7; the linkage map was constructed from an interspecific LR-26 recombinant inbred line (RIL) population derived from a cross between L. culinaris Eston and L. ervoides accession IG 72815. The QTL positions are shown with a red bar and the loci within the QTL regions are colored with blue. The green locus indicates the position of a significant marker from Bhadauria et al. (2017). Only portions of the linkage map related to the QTL positions are displayed

Box plots showing percent anthracnose severity values of the allele classes of tightly linked SNP markers to the identified QTLs on linkage groups (LG) 3 (a) and 7 (b); the QTLs were mapped using an interspecific LR-26 recombinant inbred line (RIL) population derived from a cross between L. culinaris Eston and L. ervoides accession IG 72815. The black horizontal lines in the middle of the boxes are the median values. The vertical size of the boxes represents the inter-quantile range. The upper and lower whiskers represent the minimum and maximum values of data
Identification of putative candidate genes
Scanning of the two QTL regions on the annotated reference lentil genome within a 1.5-LOD interval revealed more than 290 and 140 genes of known functions on chromosomes 3 and 7, respectively. Further analysis of these candidate genes revealed 22 genes on chromosome 3 and 26 genes on chromosome 7 that were annotated as possibly associated with plant disease resistance and/or defense-related genes. Genes within these QTL intervals that may condition anthracnose resistance include NB-ARC domain disease resistance genes, LRR receptor-like kinases (LRR-RLK), transmembrane proteins (TM), pentatricopeptide repeats proteins (PPRP), cellulose synthase proteins, ring figure proteins, serine/threonine kinase family proteins, peroxidases, ABC-transporters, and F-box proteins (Table S1).
Discussion
The absence of allelic diversity in the cultivated lentil gene pool for anthracnose race 0 resistance necessitated the introduction of resistance gene from L. ervoides which is in the tertiary gene pool of Lens. Interspecific introgression of anthracnose resistance from this source into elite cultivars could be facilitated using marker-assisted selection if there were linked markers. To detect the QTLs conditioning anthracnose resistance for race 0 and race 1 of the pathogen, we used an interspecific RIL population derived from a resistant L. ervoides accession, IG 72815 (Tullu et al. 2013). The RIL population showed significant variation in disease reaction for both races, conferred by resistance genes/alleles. A high positive correlation for disease reaction was found between race 0 and race 1, supporting the hypothesis that the resistance loci inherited from IG 72815 for both races can be co-localized. Similar results were reported for both races in another L. culinaris x L. ervoides interspecific population (Fiala et al. 2009), and in a L. ervoides intraspecific RIL population (Bhadauria et al. 2017).
The GBS-based linkage map generated for the interspecific LR-26 RIL population covered the seven chromosomes of lentil with 5455 SNP markers. The genetic linkage map spanned 3252.8 cM with an average marker density of 0.6 cM. Stange et al. (2013) reported that an increase in marker density from 5 to 1 cM could increase the power sufficiently to precisely localize and resolve closely linked QTL. Although the map size is inflated, the LR-26 genetic map developed in this study has sufficient marker density to provide adequate power for QTL mapping. Using this genetic map and the anthracnose phenotypic data of LR-26 populations, we mapped a major QTL on linkage group 3, and also on the distal end of linkage group 7. The major QTL for anthracnose resistance on chromosome 3 (qANTH-3) was derived from IG 72815, with an explained phenotypic variation ranging from 20.1 to 31.2%, conferring resistance to both races. The SNP marker previously reported in the L. ervoides genome by Bhadauria et al. (2017) for both races was located in the same region as this QTL (Fig. 2) and is quite likely the same QTL. Another QTL, with moderate effect, was detected on chromosome 7 (qANTH-7) and also conferred resistance to both races. Bhadauria et al. (2017) identified a QTL conferring resistance to race 0 on chromosome 7 using the intraspecific RIL population (LR-66) derived from race 0 and race 1 resistant L. ervoides parents (L-01-827A × IG 72815). The qANTH-7 identified in IG 72815 in this study conferred resistance to both races, possibly because accessions L-01-827A and IG 72815 carry the same race 1 resistance gene in this region of chromosome 7 and thus was not segregating in LR-66. Alternatively, this could be a different locus nearby. Murube et al. (2019) reported a co-occurring QTL conferring anthracnose resistance in common bean (Phaseolus vulgaris L.) to multiple races of C. lindemuthianum, in a region where multiple race-specific genes are found in clusters. Thus, the race 0 and race 1 resistances in accession IG 72815 could possibly be controlled by tightly clustered genes that are co-inherited.
The peak locus for qANTH-3 is 1 cM away from a large gap (21.2 cM) close to the middle of linkage group 3. This locus most likely includes the centromere; thus, the large gap interval could be due to low SNP density around the centromeric region. Underrepresentation of SNPs in pericentromeric regions were reported when using methylation-sensitive enzyme based-GBS in sorghum (Sorghum bicolor) (Patil et al. 2017). Moreover, Felderhoff et al. (2016) reported a locus spanning 48.7 Mb that includes the centromere, when mapping QTL for sorghum anthracnose resistance using GBS.
In this study we found a wealth of candidate genes that may play a role in disease resistance and plant defense-related genes in the two QTL regions. The molecular basis of disease resistance in plants is mediated through a suite of cellular receptors that perform direct detection of pathogenic molecules (reviewed by Andersen et al. 2018). This relies on the recognition of conserved pathogen-associated or microbe-associated molecular patterns (PAMPs or MAMPs) or effectors from pathogens (Martin et al. 2003). We identified candidate genes encoding for LRR-receptor-like kinase (LRR-RLK) and transmembrane proteins (TM), which are associated with the PAMPs response mechanisms, underlying the two QTL. The LRR-RLK constitutes a diverse group of proteins (also called pattern recognition receptors) allowing the cell to recognize and elicit defense responses (Torii 2004). Burt et al. (2015) reported 27 LRR-RLK related candidate genes associated within a physical region of 936.46 kb for anthracnose resistance in common bean.
We also identified a NB-ARC domain disease resistance gene under both QTL. NB-ARC class genes typically encode R genes that usually detect the pathogen and activate downstream signaling, leading to pathogen resistance (Dodds and Rathjen 2010). Receptor-like serine/threonine kinases are also associated with defense mechanisms and play a vital role in the signal transduction pathway in plants (Zhou et al. 1995). The expression of genes encoding peroxidase (Almagro et al. 2009), wall-associated receptor kinase (Delteil et al. 2016) and cellulose synthase (Douchkov et al. 2016), occurs in response to attack by pathogens, resulting in the strengthening the plant cell wall, an important first line of defense.
Genes belonging to the pentatricopeptide repeat protein (PPRP) family, which are considered resistance-related genes (Sekhwal et al. 2015), were also identified. PPRP are known to affect post-transcriptional regulations such as RNA editing, splicing and translation modification (Schmitz-Linneweber and Small 2008) and are involved in plant disease resistance (Garcia-Andrade et al. 2013). Other candidate plant defense-related genes found in the QTL regions (Table S1), include zinc‐finger proteins (Shi et al. 2014). Ogutcen et al. (2018) found copy number variation between L. ervoides and L. culinaris accessions in the coding regions of a zinc‐finger transcription factor gene.
Conclusion
The aim of this study was to identify QTL associated with anthracnose resistance in L. ervoides accession IG 72815 using an interspecific RIL population. The source of resistance in L. ervoides accession IG 72815 appears to be derived from resistance loci on chromosomes 3 and 7 for both races, an indication that resistance is possibly controlled by tightly clustered genes that are co-inherited. The SNP markers linked to these QTL will be useful in the breeding program for marker-assisted introgression of anthracnose resistance into cultivated lentil after their validation in appropriate segregating populations. Multiple classes of candidate genes that encode plant disease resistance are identified within the QTL regions that will need to be considered in follow-up validation studies.
Data availability
The raw genotypic data supporting this study are available at https:// knowpulse.usask.ca/ Geneticmap/2691115 upon request from the authors.
References
Almagro L, Ros LG, Belchi-Navarro S, Bru R, Barceló AR, Pedreno MA (2009) Class III peroxidases in plant defence reactions. J Exp Bot 60:377–390
Andersen EJ, Ali S, Byamukama E, Yen Y, Nepal MP (2018) Disease resistance mechanisms in plants. Genes (Basel) 339:1–30
Banniza S, Warale R, Menat J, Cohen-Skali A, Armstrong-Cho C, Bhadauria V (2018) The long path to understanding the host-pathogen interactions of Colletotrichum lentis on lentil. Can J Plant Pathol 40(2):199–209
Barilli E, Moral J, Aznar-Fernández T, Rubiales D (2020) Resistance to anthracnose (Colletotrichum lentis, Race 0) in Lens spp. germplasm. Agronomy 10:1799
Bhadauria V, Ramsay L, Bett KE, Banniza S (2017) QTL mapping reveals genetic determinants of fungal disease resistance in the wild lentil species Lens ervoides. Sci Rep-UK 7:3231
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120
Broman KW, Wu H, Sen S, Churchill GA (2003) R/qtl: QTL mapping in experimental crosses. Bioinformatics 19(7):889–890
Buchwaldt L, Anderson KL, Morrall RAA, Gossen BD, Bernier CC (2004) Identification of lentil germplasm resistant to Colletotrichum truncatum and characterization of two pathogen races. Phytopathology 94:236–243
Buchwaldt L, Dzananovic E, Durkin J (2018) Lentil anthracnose: epidemiology, fungicide decision support system, resistance and pathogen races. Can J Plant Pathol 40(2):189–198
Buchwaldt L, Shaikh R, Adam J, Tullu A, Slinkard A (2013) Recessive and dominant genes confer resistance to Colletotrichum truncatum in cultivated lentil. Can J Plant Pathol 35:222–231
Burt AJ, William HM, Perry G, Khanal R, Pauls KP, Kelly JD, Navabi A (2015) Candidate gene identification with SNP marker-based fine mapping of anthracnose resistance gene Co-4 in common bean. PLoS ONE 10(10):e0139450
Chen L (2018) Assessing impacts of crop-wild introgression in lentil using interspecific lens species recombinant inbred line populations. Ph.D. Dissertation, University of Saskatchewan
Chongo G, Bernier CC, Buchwaldt L (1999) Control of anthracnose in lentil using partial resistance and fungicide applications. Can J Plant Pathol 21:16–22
Coyne CJ, Kumar S, von Wettberg EJB, Marques E, Berger JD, Redden RJ, Ellis THN, Brus J, Zablatzka L, Smykal P (2020) Potential and limits of exploitation of crop wild relatives for pea, lentil, and chickpea improvement. Legume Sci 2(2):e36
Cubero JI, Pérez de la Vega M, Fratini R (2009) Origin, phylogeny, domestication and spread. In: Erskine W, Muehlbauer FJ, Sarker A, Sharma B (eds) The lentil: botany, production and uses. CAB International, Wallingford, pp 13–33
Damm U, O’Connell RJ, Groenewald JZ, Crous PW (2014) The Colletotrichum destructivum species complex hemibiotrophic pathogens of forage and field crops. Stud Mycol 79:49–84
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, McVean G, Durbin R (2011) The variant call format and VCFtools. Bioinformatics 27(15):2156–2158
Delteil A, Gobbato E, Cayrol B, Estevan J, Michel-Romiti C, Dievart A, Kroj T, Morel JB (2016) Several wall-associated kinases participate positively and negatively in basal defense against rice blast fungus. BMC Plant Biol 16:17
Dempewolf H, Baute G, Anderson J, Kilian B, Smith C, Guarino L (2017) Past and future use of wild relatives in crop breeding. Crop Sci 57:1070–1082
Dodds P, Rathjen J (2010) Plant immunity: towards an integrated view of plant-pathogen interactions. Nat Rev Genet 11:539–548
Douchkov D, Lueck S, Hensel G, Kumlehn J, Rajaraman J, Johrde A, Doblin MS, Beahan CT, Kopischke M, Fuchs R, Lipka V, Niks RE, Bulone V, Chowdhury J, Little A, Burton RA, Bacic A, Fincher GB, Schweize P (2016) The barley (Hordeum vulgare) cellulose synthase-like D2 gene (HvCslD2) mediates penetration resistance to host-adapted and nonhost isolates of the powdery mildew fungus. New Phytol 212:421–433
FAOSTAT (2017) Food and agriculture organization of the United Nations (FAO) Statistical Division. http://www.fao.org/faostat/en/#data/QC
Felderhoff TJ, McIntyre LM, Saballos A, Vermerris W (2016) Using genotyping by sequencing to map two novel anthracnose resistance loci in Sorghum bicolor. G3 6(7):1935–1946
Fiala JV, Tullu A, Banniza S, Séguin-Swartz G, Vandenberg A (2009) Interspecies transfer of resistance to anthracnose in lentil (Lens culinaris Medic.). Crop Sci 49:825–830
Garcia-Andrade J, Ramirez V, Lopez A, Vera P (2013) Mediated plastid RNA editing in plant immunity. PLoS Pathog 9:e1003713
Gela TS, Banniza S, Vandenberg A (2020) Lack of effective resistance to the virulent race of Colletotrichum lentis in Lens culinaris Medikus subsp. culinaris. Plant Gen Res Char Util 18(2):81–87
Haley CS, Knott SA (1992) A simple regression method for mapping quantitative trait loci in line crosses using flanking markers. Heredity 69(4):315–324
Kissinger G (2016) Pulse crops and sustainability: a framework to evaluate multiple benefits. http://www.fao.org/pulses
Ladizinsky G, Braun D, Goshen D, Muehlbauer F (1984) The biological species of the genus Lens L. Bot Gaz 145:253–261
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer H, Marth G, Abecasis G, Durbin R (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079
Manichaikul A, Moon JY, Sen S, Yandell BS, Broman KW (2009) A model selection approach for the identification of quantitative trait loci in experimental crosses, allowing epistasis. Genetics 181:1077–1086
Martin GB, Bogdanove AJ, Sessa G (2003) Understanding the functions of plant disease resistance proteins. Annu Rev Plant Biol 54:23–61
Morrall RAA, Pedersen EA (1990) Discovery of lentil anthracnose in Saskatchewan in 1990. Can Plant Dis Surv 71:105–106
Morrall RAA (1988) A new disease of lentil induced by Colletotrichum truncatum in Manitoba. Plant Dis 72:994
Murube E, Campa A, Ferreira JJ (2019) Integrating genetic and physical positions of the anthracnose resistance genes described in bean chromosomes Pv01 and Pv04. PLoS ONE 14(2):e0212298
Ogutcen E, Ramsay L, von Wettberg EB, Bett KE (2018) Capturing variation in Lens (Fabaceae): development and utility of an exome capture array for lentil. Appl Plant Sci 6:e01165
Patil NY, Klein RR, Williams CL, Collins SD, Knoll JE, Burrell AM, Anderson WF, Rooney WL, Klein PE (2017) Quantitative trait loci associated with anthracnose resistance in sorghum. Crop Sci 57:877–890
Podder R, Banniza S, Vandenberg A (2013) Screening of wild and cultivated lentil germplasm for resistance to Stemphylium blight. Plant Genet Res 11:26–35
Poland JA, Brown PJ, Sorrells ME, Jannink JL (2012) Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS ONE 7:e32253
Raghuvanshi RS, Singh DP (2009) Food preparation and use. In: Erskine W, Muehlbauer FJ, Sarker A, Sharma B (eds) The lentil: botany, production and uses. CABI Cambridge, Cambridge, MA, pp 408–424
Ramsay L, Koh C, Konkin D, Cook D, Penmetsa V, Dongying G, Coyne C, Humann J, Kaur S, Dolezel J, Bett KE (2019) Lens culinaris CDC Redberry genome assembly v2.0. Retrieved from https://knowpulse.usask.ca/genome-assembly/Lcu.2RBY
Rebai A (1997) Comparison of methods of regression interval mapping in QTL analysis with non-normal traits. Genet Res 65:68–74
Schmitz-Linneweber C, Small I (2008) Pentatricopeptide repeat proteins: a socket set for organelle gene expression. Trends Plant Sci 13:663–670
Sekhwal MK, Li P, Lam I, Wang X, Cloutier S, You FM (2015) Disease resistance gene analogs (RGAs) in plants. Int J Mol Sci 16:19248–19290
Shi H, Wang X, Ye T, Chen F, Deng J, Yang P, Zhang Y, Chan Z (2014) The Cysteine2/Histidine2-Type transcription factor ZINC FINGER OF ARABIDOPSISTHALIANA6 modulates biotic and abiotic stress responses by activating salicylic acid-related genes and C-REPEAT-BINDING FACTOR genes in Arabidopsis. Plant Physiol 165:1367–1379
Slinkard AE (1981) Cultivar description: Eston lentil. Can J Plant Sci 61:733–734
Stange M, Utz HF, Schrag TA, Melchinger AE, Würschum T (2013) High-density genotyping: an overkill for QTL mapping? Lessons learned from a case study in maize and simulations. Theor Appl Genet 126:2563–2574
Tanksley S, McCouch S (1997) Seed banks and molecular maps: unlocking genetic potential from the wild. Science 277:1063–1066
Taylor J, Butler D (2017) R package ASMap: efficient genetic linkage map construction and diagnosis. J Stat Softw 79(6):1–29
Torii KU (2004) Leucine-rich repeat receptor kinases in plants: structure, function, and signal transduction pathways. Int Rev Cytol 234:1–46
Tullu A, Banniza S, Tar’an B, Warkentin T, Vandenberg A (2010) Sources of resistance to ascochyta blight in wild species of lentil (Lens culinaris Medik.). Genet Resour Crop Evol 57:1053–1063
Tullu A, Bett K, Banniza S, Vail S, Vandenberg A (2013) Widening the genetic base of cultivated lentil through hybridization of Lens culinaris “Eston” and L. ervoides accession IG 72815. Can J Plant Sci 93:1037–1047
Tullu A, Buchwaldt L, Lulsdorf M, Banniza S, Barlow B, Slinkard AE, Sarker A, Tar’an B, Warkentin T, Vandenberg A (2006) Sources of resistance to anthracnose (Colletotrichum truncatum) in wild Lens species. Genet Res Crop Evol 53:111–119
Tullu A, Buchwaldt L, Warkentin T, Tar’an B, Vandenberg A (2003) Genetics of resistance to anthracnose and identification of AFLP and RAPD markers linked to the resistance gene in PI 320937 germplasm of lentil (Lens culinaris Medikus). Theor Appl Genet 106:428–234
Van Oss H, Aron Y, Ladizinsky G (1997) Chloroplast DNA variation and evolution in the genus Lens Mill. Theor Appl Genet 94:452–457
Wong MML, Gujaria-Verma N, Ramsay L, Yuan HY, Caron C, Diapari M, Vandenberg A, Bett KE (2015) Classification and characterization of species within the genus Lens using genotyping-by-sequencing (GBS). PLoS ONE 10(3):e0122025
Zeng ZB (1994) Precision mapping of quantitative trait loci. Genetics 136(4):1457–1468
Zhou J, Loh YT, Bressan RA, Martin GB (1995) The tomato gene Pti1 encodes a serine/threonine kinase that is phosphorylated by Pto and is involved in the hypersensitive response. Cell 83:925–935
Acknowledgements
The authors gratefully acknowledge funding from the Natural Sciences and Engineering Research Council of Canada (NSERC) Industrial Research Chair Program, the Saskatchewan Pulse Growers, and the University of Saskatchewan. We are also thankful for the technical assistance of the bioinformatics, molecular and pathology lab staff of the Pulse Breeding and Genetics group at the University of Saskatchewan.
Author information
Authors and Affiliations
Contributions
TSG generated phenotypic data, preformed data analyses and wrote the manuscript; LC performed the genotyping of the mapping population; CC carried out SNP calling; CSK constructed the genetic map, AV and KEB conceived the experiment and critically reviewed the manuscript. All authors read and approved the final version.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Gela, T.S., Koh, C.S., Caron, C.T. et al. QTL mapping of lentil anthracnose (Colletotrichum lentis) resistance from Lens ervoides accession IG 72815 in an interspecific RIL population. Euphytica 217, 70 (2021). https://doi.org/10.1007/s10681-021-02804-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10681-021-02804-0