
Abstract
Global efforts are underway to develop staple crops with improved levels of provitamin A carotenoids to help combat dietary vitamin A deficiency, which is widespread among resource-poor farmers in the developing world. As a staple crop for more than 500 million people in sub-Saharan Africa, cassava (Manihot esculenta Crantz) enriched with provitamin A carotenoids could have immense nutritional impact. To this effort, a genome-wide association study (GWAS) was conducted with the aim of identifying genomic regions controlling variation for carotenoid concentration in cassava as a basis for undertaking systematic genetic improvement for carotenoid content. A high throughput genotyping-by-sequencing platform was used to genotype a diverse association panel of 591 genotypes and identified 179,310 single nucleotide polymorphisms (SNPs) distributed across the cassava genome. A genomic region stretching 1.37 Mb on chromosome 1 was identified with four SNPs significantly associated with carotenoid content in cassava. A survey of cassava genome sequence v6.1 positioned these SNPs in the vicinity of Manes.01G124200.1 locus, which is a known gene responsible for increase in accumulation of provitamin A carotenoids in cassava roots. This study provides one of the pioneer insights into the practical application of GWAS for dissecting the genetic basis of nutritional traits in cassava. Findings presented herein offer practical grounds for improving carotenoid content in cassava through marker-assisted breeding and genomic selection.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
As the world population size approaches the nine billion mark, cassava (Manihot esculenta Crantz) is projected to be a cornerstone on which to rest aspirations for meeting increased calorie demands in sub-Saharan Africa (SSA), which are currently experiencing the highest population growth rates in the world (Edgerton 2009; Burns et al. 2010; Tan 2015). The starchy root crop is grown and consumed widely in tropical regions of Africa, Asia and Latin America, where it dependably provides household food security in resource-poor farming communities (Monfreda et al. 2008; Salvador et al. 2014). Globally, at least 800 million people derive the bulk of their dietary energy from cassava on a daily basis and over 500 million of these people live in SSA (FAOSTAT 2009; Montagnac et al. 2009; Burns et al. 2010). In Uganda, cassava is the second most consumed crop after banana. However, more than 90 % of the edible portion of cassava roots consists of carbohydrates (Montagnac et al. 2009), making diets that heavily depend on cassava to be deficient in essential micronutrients such as vitamin A, iron and zinc. In particular, vitamin A deficiency (VAD) has remained a widespread nutritional challenge (WHO 2009; Manson et al. 2014).
Consequently, people that depend on cassava for food are vulnerable to VAD, which is associated with corneal blindness, reduced immunity to preventable diseases, stunted growth, anaemia and reduced capacity for physical activity (Gegios et al. 2010; Stephenson et al. 2010). It was in response to this distinct challenge that a special breeding agenda, referred to as biofortification, was initiated in the early 2000s under the HarvestPlus project to develop cassava varieties enriched with provitamin A carotenoids (Pfeiffer and McClafferty 2007). In practice, consumption of provitamin A cassava can be considered a home-based therapy, as it is expected to minimise VAD that is widespread among resource-poor farmers that primarily depend on cassava (Mayer et al. 2008; Talsma et al. 2013).
However, the intrinsic heterozygous nature, long breeding cycles and high sensitivity to environmental variations make cassava a difficult crop to breed conventionally (Ceballos et al. 2004, 2015). Nonetheless, recent advances in molecular breeding technologies have proved to be useful for increasing genetic gains for complex traits in crops such as wheat (Poland et al. 2012), maize (Ersoz et al. 2009; Crossa et al. 2013) and rice (Yuan-yuan et al. 2014) and a replica of such technologies is expected to enhance genetic improvement of cassava (Oliveira et al. 2012). Indeed, a classic example for this was the pioneer marker-assisted introgression of cassava mosaic disease resistance into Latin American cassava germplasm prior to its introduction into Africa (Okogbenin et al. 2007). This is a classical form of marker-assisted selection (MAS), in which either single genes or quantitative trait loci (QTL) were successfully introgressed. In the case of cassava carotenoids, patterns of phenotypic segregation appear to suggest that the trait is controlled by more than one gene (Akinwale et al. 2010). Efforts that can pinpoint the allelic basis of such variability would, for example, enhance chances of using gene-based selection to facilitate the rapid recurrent selection scheme that has already been demonstrated for carotenoid content in cassava (Ceballos et al. 2013).
Recent advances in next generation sequencing (NGS) technologies have made linkage disequilibrium (LD)-based genome-wide association study (GWAS) a favourable tool for mapping complex traits in higher plants (Davey et al. 2011). It is now practical to use high-density genotyping platforms such as genotyping-by-sequencing (GBS) to quickly generate millions of marker data points that are distributed throughout a genome for purposes of facilitating genetic improvement in plants (He et al. 2014). For example, Owens et al. (2014) and Suwarno et al. (2015) have used these NGS platforms to identify polymorphisms linked to increased carotenoid contents in maize, which certainly provides a foundation for systematic provitamin A biofortification. With more than 97 % of the cassava genome sequenced (Prochnik et al. 2012), it is now possible to use GBS (Elshire et al. 2011) and GWAS (Oliveira et al. 2012) to precisely anchor single nucleotide polymorphisms (SNPs) linked to traits of interest. Indeed, these platforms make it easy to infer functions of such polymorphisms based on annotations of genomic regions where they are positioned. Rabbi et al. (2014) used SNP data generated from the GBS platform to develop a high-resolution genetic map that identified genomic regions of cassava controlling resistance to cassava mosaic geminiviruses.
It is against this background that the national cassava breeding programme in Uganda recently initiated a breeding objective tailored towards development of provitamin A carotenoid content in cassava by harnessing advancements in genomics, notably the GBS technology, and diverse carotene-rich genetic resources. Specifically, the current study was conducted to identify genomic regions and SNPs linked to natural variations for carotenoid content in cassava as a basis for more systematic carotenoid biofortification in the crop.
Materials and methods
Genotypes
A panel of 655 genotypes was considered for this study. These genotypes included an array of S1 and S2 partial inbreds generated from eight S0 progenitors of diverse genetic background (Table 1). S0 progenitors were carotene-rich lines selected from a set of clones previously introduced from the International Center for Tropical Agriculture (CIAT) and International Institute for Tropical Agriculture (IITA) for breeding in Uganda (Esuma et al. 2012). S0 progenitors were self-pollinated to generate S1 progeny during the 2009–2010 cropping season. Thirty-nine S1 clones with the ability to flower were planted in a crossing block in August 2011 and self-pollinated to generate S2 genotypes. In all cases, self-pollinations were performed following the standard procedure described by Kawano (1980).
Phenotyping
Clonal trials for the 665 association panel were conducted between June 2014 and May 2015 at two locations in Uganda: Abi Zonal Agricultural Research and Development Institute (Abi-ZARDI) and National Crops Resources Research Institute (NaCRRI), which have different agro-ecological characteristics. NaCRRI is located within the Lake Victoria crescent at 32°37′36.0″E and 0°31′13.7″N, 1134 m above sea level, while Abi-ZARDI is located within north-western savannah grasslands at 31°1′28.4″E and 2°36′33.3″N, 1060 m above sea level. At each site, trials were laid out in an incomplete block design (Clewer and Scarisbrick 2001) with two replications. Plots in a block consisted of single rows of three plants each. Planting was done at a spacing of 1 × 1 m. Trials were entirely rain fed and all agronomic practices including weed management were strictly followed. Total carotenoid content (TCC) of roots was measured at 12 months after planting using the iCheck analytical kit developed by BioAnalyt Laboratory, Germany (http://www.bioanalyt.com).
Briefly, three roots (with the ability to provide ≥250 g parenchyma tissue) were processed for measurement of TCC, each root being a random selection from each of the three plants in a plot. The selected roots were peeled, washed under running water, dried with a paper towel and cut longitudinally into quarters so that the opposite quarters were pooled, chopped into small pieces and homogenised. About 5 g of the homogenous root sample was pounded and ground into a smooth and fine paste using a mortar and pestle. To aid grinding of the sample, 20 ml of distilled water was added gradually and the resulting solution transferred into a 50 ml calibrated tube. The tube content was shaken thoroughly and 0.4 ml of the solution injected into the iEx™ CAROTENE vial using the syringe and needle provided with the kit. Vials were placed on a solid surface for approximately 5 min, shaken again and allowed to stand until two solution phases appeared inside the vial: a clear upper phase and a turbid lower phase. At this point, the absorbance of the vial content (the upper solution phase) was measured using the iCheck™ CAROTENE device. TCC was calculated as:
where Vs = volume of solution transferred to the tube, Ws = weight of sample and A = absorbance of the iEx™ CAROTENE vial content at a wavelength of 450 nm. Each sample was extracted and measured for TCC once. All procedures for carotenoid quantification were performed in a dark room and harvested roots were analysed for TCC within 12 h. Root flesh colour (RFC) was also scored for all genotypes using visual inspection following the standard colour scale as described by (Sánchez et al. 2006).
Genotyping
Total genomic DNA was extracted from newly expanded plant leaves according to the procedure described by Dellaporta et al. (1983). The GBS platform described by Elshire et al. (2011) was used to genotype the association panel. Briefly, restriction enzyme ApeKI, which is a type II restriction endonuclease that recognises a degenerate 5 bp sequence (GCWGC, where W is A or T), was used to digest DNA samples. ApeKI was selected as the restriction enzyme because it has partial sensitivity to DNA methylation and cuts genomic DNA at low frequency, thus avoiding repetitive genomic regions and enhancing adapter ligation to insert DNA (Peterson et al. 2014). GBS sequencing libraries (95-plex) were prepared by ligating the digested DNA to barcodes (unique nucleotide adapters) followed by standard polymerase chain reaction. Libraries were sequenced using 1-lane 86 bp reads using the Illumina HiSeq 2000. Sequence reads from different genotypes were de-convoluted using barcodes and aligned to the cassava reference genome sequence v6.1 (Glaubitz et al. 2014). The TASSEL-GBS pipeline was used to make SNP calls following the bioinformatics procedures described in Elshire et al. (2011) and Glaubitz et al. (2014).
Genotypic data were further processed to ensure high quality for subsequent analyses. The dataset originally had close to 70 % missing data points, which necessitated imputation of the non-genotyped markers, as described in earlier studies (Romay et al. 2013; Swarts et al. 2014). To achieve this, indels were first removed from the raw sequence data using TASSEL v5.2.9 (Bradbury et al. 2007) and the cleaned dataset imputed using Beagle software v4.0 based on an algorithm which searches for the closest neighbour in small SNP windows across the entire genome (Browning and Browning 2013; Swarts et al. 2014). To remove non-segregating and uninformative sites, the imputed data were further filtered at the following thresholds: minor allele frequency (MAF) = 0.01, minimum count of genotypes = 50 and minimum heterozygous proportion = 0.01. This filtered dataset represented a reduced marker set comprising 179,310 SNPs with a robust MAF desirable for estimating population structure and kinship, both of which were important for subsequent statistical analyses.
Statistical analysis
Out of the 655 individuals initially considered for this study, 591 genotypes had both phenotypic and genotypic data and were used for subsequent statistical analyses. A mixed linear model (MLM) fitted across environments was used to conduct analysis of variance (ANOVA) using the lme4 package of the R software (R Development Core Team 2010; Vazquez et al. 2010). The model used was:
where Yijk = phenotypic value, μ = overall phenotypic mean, β i = effect of environment i, R ij = effect of block j in environment i, G k = effect of genotype k, (β i , × G k ) = effect of interaction between environment i and genotype k and e ijkm = residual. Both the phenotypic means and best linear unbiased predictions (BLUPs) extracted from the MLM were used for genome-wide association (GWA) testing to benefit from the additional power that this might offer.
Unless stated, all statistical analyses of the genotypic data were performed using TASSEL v5.2.9 (Bradbury et al. 2007). Principal component analysis (PCA) through eigenvalue decomposition of the covariance matrix was performed using the filtered SNP set to infer population structure. The first two axes of the PCA were used to draw a scatter plot to visualise genetic differentiation among genotypes. In execution of GWA analysis, the first three principal components constituted the population structure. A kinship matrix was constructed using the scaled_IBS method (IBS = identical by state). This method produces a matrix that is scaled to give a reasonable estimate of the additive genetic variance, which effectively controls statistical errors arising from population structure in a MLM for the analysis. The kinship matrix was calculated according to Bishop and Williamson (1990):
where IBS1 = state in which two genotypes share one allele at a locus and IBS2 = state in which two genotypes share two alleles at a locus.
Genome-wide and chromosome-wise LD were assessed based on adjacent pairwise r 2 values (the squared correlation coefficients among alleles at two adjacent SNP markers) and physical distances among these SNPs (Remington et al. 2001). LD was estimated using the unimputed dataset consisting of 194,322 SNPs, considering only markers with <30 % missing data and MAF >0.05. Average pairwise distances in which LD decayed at r 2 = 0.2 and r 2 = 0.1 were then calculated based on the model, whereby the expected value (E) of r 2 was:
where r 2 = squared correlation coefficient, n = sample size and C = model coefficient for the distance variable (Hill and Weir 1988).
After removing monomorphic and low-quality SNPs, a total of 179,310 SNPs were available for the 591 member association panel. The MLM was used for testing individual SNP-based associations, following the correlation/trend method described by Weir (2008). The method was chosen for statistical analyses due to its ability to achieve consistently good control over false positives while yielding the highest power among other structure correction methods (Wang et al. 2012). The MLM is illustrated as:
where Y = phenotypic response of the dependent variable (means or BLUPs of TCC), SNP = SNP marker (fixed effect), β = fixed effect model coefficient for SNP, PC = first three components of PCA (which accounted for 97.6 % variation) to present population structure (fixed effect), α = fixed effect model coefficient for PC (population structure), K = kinship matrix (random effect) and ε = residual. Population structure was incorporated into this model to correct for their confounding effects on marker-trait associations, thus controlling false positives (Li et al. 2014).
Evaluations of the association mapping model were based on the quantile–quantile (Q–Q) plot, which is a plot of observed −log10 P values versus expected −log10 P values under the null hypothesis that there is no association between a SNP and the phenotype. The approach used for identifying GWA signals was based entirely on the smallest P values obtained from the MLM, where SNPs were ranked based on ascending order of their P values. SNPs with P values less than the 5 % Bonferroni threshold were considered to be significantly associated with phenotypes. Furthermore, chromosome-wise association signals were visualised from Manhattan plots generated using the qqman package of R software (Turner 2014). We browsed the cassava reference genome sequence v6.1 using phytozome v11.0 (Goodstein et al. 2012) to search for model genes in the proximities of significant SNPs. To overcome issues of non-collinearity that may exist within this genome version (given it is only partially complete), broader intervals (up to 0.5 Mb) were considered while searching for pathway genes in the vicinity of SNPs showing significant associations.
Results
Phenotypic variability and correlations
For the 591 genotypes considered for statistical analyses, average TCC was 4.9 µg g−1, varying from 0 to 13.45 µg g−1 on a fresh weight basis, which presented considerable variability for carotenoid content. RFC varied from 1 (white) to 6 (deep yellow). Distribution of the genotypes based on RFC revealed that 135 clones were white-fleshed, 37 light cream, 99 cream, 134 light yellow, 129 yellow and 57 deep yellow. ANOVA showed significant genotypic effects for TCC, but environmental effects were non-significant (Supplementary Table 1). Broad-sense heritability was high for TCC (0.91). Phenotypic values and BLUPs for TCC showed a bimodal distribution (Fig. 1). The Pearson correlation coefficient between TCC and RFC was high, with r = 0.84 (P ≤ 0.01).

Scatter plot and histograms of total carotenoid content (TCC) versus best linear unbiased predictions (BLUPs) of TCC. Colour 1-WT white, 2-LC light cream, 3-CM cream, 4-LY light yellow, 5-YL yellow, 6-DY deep yellow, red lines on histograms indicate means
Marker coverage and missing data
A total of 635 247 SNP markers were used to genotype the panel of 655 clones. Overall, there was a low SNP call rate, with an average proportion of missing data of 71 % (Supplementary Table 2). The average SNP call rate per sample was 28.2 %, varying from 1.3 to 81.4 %. Following removal of monomorphic markers and imputation of the genotype subset, 179 310 SNPs were selected and used for subsequent analyses. These SNPs were distributed along the 18 chromosomes and two linkage groups of unanchored scaffolds of the cassava genome sequence v6.1. When further filtering of the GBS data was done to remove genotypes missing phenotypes, a total of 591 genotypes were left for the association testing. A comparison of the imputed GBS data with results from the unprocessed GBS SNP data for the 591 member association panel showed a median discrepancy rate of 8.2 % for all calls. When heterozygous calls were excluded, the median error rate was reduced to 4.3 %, which highlighted high accuracy of imputation. The imputed data were therefore used to perform GWA analysis.
Population structure, allele frequency and linkage disequilibrium
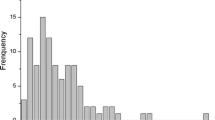
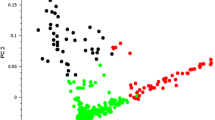
Cassava genotypes used in this study were drawn from breeding populations developed using eight parental lines sourced from either IITA or CIAT; thus they presented a set of genotypes from diverse genetic backgrounds, which would be a possible source of population structure. PCA was used to describe the population structure for the association panel for this study. Grouping of genotypes along the axis of the first two principal components scattered genotypes around the centre, without clear-cut differentiation between them. However, classifying genotypes according to their origin revealed some level of genetic differentiation, with the IITA group drifting to the left along the first principal component axis (Fig. 2). When the PCA was examined using the GAPIT programme (Lipka et al. 2012) for analysis of population structure, the clustering pattern suggested some structure within the population studied (Supplementary Fig. 1). This apparent genetic differentiation provided the basis for using both PCA and the kinship matrix in the MLM for association analysis, which was an important strategy to minimise any false positives in the mapping result. Overall, 82 % of the SNPs typed in the entire population had MAF <0.01 (Fig. 3), with more than 71 % of the alleles shared between the CIAT and IITA accessions. The average physical distance between pairs of markers was 23.4 kb and average genome-wide LD (r 2) obtained based on adjacent pairs of markers was 0.29. Genome-wide LD decay was 1.32 kb at r 2 = 0.2 and 3.21 kb at r 2 = 0.1 (Supplementary Fig. 2).

Scatter plot of 591 genotypes based on principal component analysis. PC1 principal component 1, PC2 principal component 2, CIAT International Centre for Tropical Agriculture, IITA International Institute for Tropical Agriculture

Distribution of minor allele frequency (MAF) based on unfiltered genotyping by sequencing data
Association results
Based on the MLM analysis for TCC BLUPs, a strong GWA signal was detected (Fig. 4) and a chromosome-wise analysis mapped the signal to chromosome 1 of the cassava genome (Fig. 5). Within this peak, four SNPs had P values smaller than the 5 % Bonferroni threshold for genome-wide significance. This association signal was consistent for analyses based on TCC means (Supplementary Fig. 3). When the association tests were performed using RFC as a phenotype, an association hit similar to that obtained for BLUPs and TCC means was detected (Supplementary Fig. 4). This particular result corroborated the strong positive association between RFC and carotenoid content. Manhattan plots consistently reflected these results. In all these cases, the same set of SNPs was identified to show significant association signals. The four SNPs with P values below the 5 % Bonferroni correction threshold were located within a stretch of 1.37 Mb on chromosome 1 (Fig. 6). The MLM analyses based on both BLUPs and TCC means indicated highest −log10(P) for marker S1_24661940 (P = 4.4E−08) followed by marker S1_24654776 (P = 1.3E−08). These two SNPs accounted for the highest phenotypic variance (9 %) for TCC and were separated by a distance of 0.01 Mb (Table 2). Meanwhile, marker S-1_23297833 had the least −log10(P) and a low phenotypic variance (7 %) for TCC.

Quantile-quantile plots for diagnosis of association signals based on best linear unbiased predictions for total carotenoid content

Manhattan plots for genome-wide diagnosis of association signals based on best linear unbiased predictions for total carotenoid content. P and Q are linkage groups containing unanchored scaffolds in the cassava genome sequence v6.1. Horizontal blue and red lines are the suggestive and 5 % Bonferroni threshold lines, respectively

Manhattan plot for genome-wide diagnosis of association for chromosome 1, zooming out the region with association signal for total carotenoid content. Horizontal blue and red lines are the suggestive and 5 % Bonferroni threshold lines, respectively
Reference was made to the cassava genome sequence v6.1 in Phytozome v10.3 (Goodstein et al. 2012) to identify annotated genes within a distance of ± 0.5 Mb of the genomic region occupied by the significant SNPs. Manes.01G124200.1, which is a phytoene synthase (PSY) gene known to increase accumulation of provitamin A carotenoid in cassava roots, was found within the genomic region occupied by significant SNPs. The genomic size of this gene is 3.3 kb stretching from 24,153,420 to 24,156,720 bp positions and falls within the 1.37 Mb portion occupied by association hit for TCC on chromosome 1. Functional annotation of this gene relates its biological activity to the enzyme PSY, which is strongly linked to carotenoid biosynthesis in plants (Goodstein et al. 2012). SNPs S1_23297833 and S1_24601429 were the significant markers that flanked Manes.01G124200.1 upstream and downstream, respectively. However, SNPs with the highest association significance for TCC were furthest downstream of the phytoene synthase locus.
Further reference was made to version 2.0 of the cassava haplotype map being constructed in the Buckler Laboratory at Cornell University, USA (unpublished data, Ramu Punna), for functional annotation of the SNPs significantly associated with TCC. Sequences of cassava genotypes rich in carotenoid content (yellow-fleshed genotypes with TCC >5 g µg−1) were compared to those of genotypes with negligible carotenoid content (white-fleshed genotypes with TCC <1 g µg−1), following an alignment in the Mega software. Amino acid lysine in a conserved region of S1_24601431 of white-fleshed genotypes was substituted by threonine in yellow-fleshed genotypes. SNP S1_24601431 in the haplotype map co-segregated with TCC in a similar pattern to that of S1_24601429 from the GBS data. Amino acid substitutions at the other three significant SNPs were non-informative with regard to carotenoid biosynthesis.
Discussion
Agricultural production across the world is being challenged by a rapid human population growth, limited arable land and adverse climate changes, which call for greater efforts to optimise the use of available resources needed to balance increased food production with reduced environmental effects. On their part, plant breeders could benefit from application of advanced genomic tools to discover and use functional genetic variations for crop improvement (Moose and Mumm 2008). Fortunately, diverse cassava genetic resources exist across the tropics for economically important traits (Nassar 2003; Nassar et al. 2007). Such useful genetic variation could be explored through GWAS to isolate and improve agriculturally important traits in a crop that will certainly provide food to this rapidly increasing human population (Ceballos et al. 2015). Accordingly, this study focused on carotenoid content, a trait of immense value for diets in SSA. Thus, a GWA testing with 179,310 SNP markers and 591 cassava partial inbreds was conducted with the view of understanding genetic causes of natural variation for carotenoid content in cassava. Such knowledge would provide a framework for undertaking more systematic genetic improvement of cassava for increasing levels of carotenoid content in roots.
It suffices to note that one serious consideration in defining an association panel is the extent of LD, which can greatly influence GWAS results because larger LD blocks and a slower rate of LD decay generally result in lower mapping resolutions (Grady et al. 2011). For example, Romay et al. (2013) observed more rapid LD decay in tropical germplasm of maize compared to that in temperate germplasm, which means a higher resolution map would be achieved by using the tropical germplasm. In the current study, two broad accession groups (originating from CIAT and IITA) showed comparable LD decay, implying that similar mapping results could be achieved with one or a combination of these sets of genotypes.
Four SNPs (S1_23297833, S1_24601429, S1_24654776 and S1_24661940) with GWA significance for TCC were identified to be located within a single major QTL. These polymorphisms were located in the vicinity of a previously characterised PSY gene with functions linked to carotenoid biosynthesis in cassava. PSY2-Y-2 was reported to increase accumulation of provitamin A carotenoids (Welsch et al. 2010) and the same gene family is known to regulate carotenoid accumulation in rice (Beyer et al. 2002), maize (Fu et al. 2010) and durum wheat (Giuliano 2014). According to Welsch et al. (2010), a SNP in PSY2-Y-2 gene co-segregated with high carotenoid content in cassava roots. This polymorphism resulted in a single amino acid change in a highly conserved region of the protein which, when tested in Escherichia coli, resulted in increased catalytic activity. Findings of the current study strongly corroborate the earlier reports on carotenoid genes in cassava by Welsch et al. (2010). It is also very likely that additional genetic variants that explain natural variation for carotenoid content in the crop exist around this genomic region. Based on the population used in this study, carotenoid accumulation in cassava roots appears to be under the control of a single gene with several allelic variants, which share strong positional relationship with a previously characterised PSY gene. Essentially, the current study strongly supports previous reports that suggested carotenoid content in cassava roots as a qualitative trait.
The GWA hits for TCC concentrated around the region occupied by Manes.01G124200.1, but the most significant SNPs within the hit were located furthest from this carotenoid gene. This apparent clustering of the significant SNPs could be a manifestation of more than one QTL for TCC within the association region, suggesting the possibility of discovering additional genes controlling accumulation of carotenoid content in cassava. Welsch et al. (2010) identified provitamin A carotenoid polymorphisms using a bi-parental mapping population of 164 individuals developed from two landrace genotypes from Brazil. An important feature of bi-parental mapping populations is that alleles segregating in progeny are limited to allelic diversity in the two parents (Semagn et al. 2010). In the current study, a panel of diverse germplasm of both African and Latin American origins was used to survey polymorphisms linked to TCC across the cassava genome. This approach may have presented an opportunity for discovering additional polymorphisms that could furthermore explain the extent of phenotypic variation for carotenoid content. It is possible that the diverse genetic material assayed in this study increased the frequency and chances of detecting novel genetic variants explaining the global phenotypic diversity for TCC (Korte and Farlow 2013).
Indeed, substitution of lysine in the white-fleshed genotypes by threonine in the yellow-fleshed genotypes at S1_24601429 strongly suggests the metabolic role of this particular SNP in carotenoid biosynthesis. Threonine has been reported as a substance encoded by PSY1 in all maize varieties that produce yellow endosperm (rich in provitamin A carotenoids) (Shumskaya et al. 2012; Shumskaya and Wurtzel 2013). A variant of threonine expressed in golden rice was also attributed to enzyme PSY in carotenoid biosynthesis (Paine et al. 2005). Thus, it can be postulated that SNP S1_24601429 is a component of a PSY gene previously reported to increase accumulation of carotenoid content in cassava roots (Welsch et al. 2010). It can further be suggested that more than one allelic variation influencing carotenoid accumulation in roots exist within this locus. For example, the allelic polymorphism in the conserved region of the PSY locus reported by Welsch et al. (2010) resulted into change from alanine (in the white-fleshed genotypes) to aspartic acid (in the yellow-fleshed genotypes), different from that found in this study where lysine was substituted to threonine. As an improved version of the whole genome sequence of cassava becomes available in future, detailed biological annotations of these SNPs and their use for systematic cassava biofortification will become more apparent.
The heterozygous nature and large genome size of cassava are important factors to consider when applying NGS technologies to study the crop (Oliveira et al. 2012; Ceballos et al. 2015). Version 6.1 of the cassava genome assembly is approximately 582.25 Mb in length with a total of 33,033 loci containing protein-coding transcripts arranged on 18 chromosomes and 2001 scaffolds that have not been anchored onto specific chromosomes (Goodstein et al. 2012; Prochnik et al. 2012). To achieve reasonable coverage of such a large genome through GBS, a large number of SNPs distributed throughout the genome would be required (Deschamps et al. 2012). In this study, 635,247 SNPs were used to genotype 591 clones. Unprocessed GBS data showed more than 70 % missing data points. After filtering to remove non-informative SNPs, 179,310 markers were used for GWA analysis, which may represent low genome coverage and thus reduced the chances of capturing more polymorphisms with statistical significance. As observed in barley (Pasam et al. 2012), SNPs in association testing commonly explain a low percentage of genetic trait variation (<4 %) compared to QTL effects (as high as 47 %) detected through bi-parental mapping (Abdel-Haleem et al. 2010), which essentially limits the ability of GWAS to detect association signals with small effects. Efforts are on-going, in collaboration with the Next Generation Cassava Breeding Project (www.nextgencassava.org), to develop genomic tools to enhance molecular breeding of cassava. Before long, such resources could make GWAS a more powerful tool for dissecting genetic architecture of important agronomic and quality traits of cassava that have been too complex to exploit through conventional breeding methods (Ceballos et al. 2012, 2015).
In conclusion, this study was one of the pioneering efforts to apply NGS technologies to elucidate genetics of cassava at molecular level and has indeed proved that GWAS is a powerful tool for unravelling genetic factors linked to natural variations for important traits in cassava. The GWAS identified four SNPs strongly linked to carotenoid content in cassava, located within the vicinity of Manes.01G124200.1, a gene known to increase accumulation of TCC in roots. These SNPs linked to TCC could provide a refined insight into genetic architecture of carotenoid content and offer a practical basis for developing cassava varieties with high levels of provitamin A carotenoids. However, further efforts are required to sequence the genomic region containing the significant association signal in order to uncover useful sequence information that can underpin metabolic roles of the associated SNPs with regard to accumulation of TCC in cassava roots. Such information will enable, for example, use of the SNPs as fixed effects when implementing genomic selection for improving cassava for carotenoid content in genetic backgrounds of relevant agronomic traits like root dry matter content, which is consistently negatively correlated with TCC in African cassava germplasm (Njoku et al. 2011, 2015; Esuma et al. 2016). Collectively, these strategies would enhance the chances of developing cassava varieties with increased levels of provitamin A carotenoids, which, when deployed to farmers, could sustainably reduce VAD that is currently prevalent where the crop is a major staple.
References
Abdel-Haleem H, Bowman J, Giroux M, Kanazin V, Talbert H, Surber L, Blake T (2010) Quantitative trait loci of acid detergent fiber and grain chemical composition in hulled x hull-less barley. Euphytica 172:405–418
Akinwale MG, Aladesanmwa RD, Akinyele BO, Dixon AGO, Odiyi AC (2010) Inheritance of ß-carotene in cassava (Manihot esculenta Crantz). Int J Genet Mol Biol 2:198–201
Beyer P, Al-Babili S, Ye X, Lucca P, Schaub P, Welsch R, Potrykus I (2002) Golden rice: introducing the beta-carotene biosynthesis pathway into rice endosperm by genetic engineering to defeat vitamin A deficiency. J Nutr 132:506–510
Bishop DT, Williamson JA (1990) The power of identity-by-state linkage methods of analysis. Am J Hum Genet 46:254–265
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Browning BL, Browning SR (2013) Improving the accuracy and efficiency of identity by descent detection in population data. Genetics 194:459–471
Burns A, Gleadow R, Cliff J, Zacarias A, Cavagnaro T (2010) Cassava: the drought, war and famine crop in a changing world. Sustainability 2:3572–3607
Ceballos H, Iglesias C, Pérez JC, Dixon AGO (2004) Cassava breeding: opportunities and challenges. Plant Mol Biol 56:503–516
Ceballos H, Kulakow P, Hershey C (2012) Cassava breeding: current status, bottlenecks and the potential of biotechnology tools. Trop Plant Biol 5:73–87
Ceballos H, Morante N, Sánchez T et al (2013) Rapid cycling recurrent selection for increased carotenoids content in cassava roots. Crop Sci 53:23–42
Ceballos H, Kawuki RS, Gracen VE, Yencho GC, Hershey CH (2015) Conventional breeding, marker-assisted selection, genomic selection and inbreeding in clonally propagated crops: a case study for cassava. Theor Appl Genet 128:1647–1667
Clewer AG, Scarisbrick DH (2001) Practical statistics and experimental design for plant crop science. Wiley, Chichester
Crossa J, Beyene Y, Kassa S et al (2013) Genomic prediction in maize breeding populations with genotyping-by-sequencing. G3—Genes Genom Genet 3:1903–1926
Davey JW, Hohenlohe PA, Etter PD, Boone JQ, Catchen JM, Blaxter ML (2011) Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat Rev Genet 12:499–510
Dellaporta S, Wood J, Hicks J (1983) A plant DNA minipreparation: version II. Plant Mol Biol Rep 1:19–21
Deschamps S, Llaca V, May GD (2012) Genotyping-by-sequencing in plants. Biology 1:460–483
Edgerton MD (2009) Increasing crop productivity to meet global needs for feed, food, and fuel. Plant Physiol 149:7–13
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PloS One 6:e19379
Ersoz ES, Yu J, Buckler ES (2009) Applications of linkage disequilibrium and association mapping in maize. In: Kritz AL, Larkins BA (eds) Molecular genetic approaches to maize improvement, vol 63. Springer, Berlin, pp 173–190
Esuma W, Rubaihayo P, Pariyo A et al (2012) Genetic diversity of provitamin A cassava in Uganda. J Plant Stud 1:60–71
Esuma W, Kawuki SR, Herselman L, Labuschagne M (2016) Stability and genotype by environment interaction of provitamin A carotenoid and dry matter content in cassava in Uganda. Breed Sci. doi:10.1270/jsbbs.16004
FAOSTAT (2009) Why cassava? http://www.fao.org/ag/AGP/agpc/gcds/index_en. Accessed on 4 Jan, 2016
Fu Z, Yan J, Zheng Y, Warburton ML, Crouch JH, Li JS (2010) Nucleotide diversity and molecular evolution of the PSY1 gene in Zea mays compared to some other grass species. Theor Appl Genet 120:709–720
Gegios A, Amthor R, Maziya-Dixon B et al (2010) Children consuming cassava as a staple food are at risk for inadequate zinc, iron, and vitamin A intake. Plant Foods Hum Nutr 65:64–70
Giuliano G (2014) Plant carotenoids: genomics meets multi-gene engineering. Curr Opin Plant Biol 19:111–117
Glaubitz JC, Casstevens TM, Lu F, Harriman J, Elshire RJ, Sun Q, Buckler ES (2014) TASSEL-GBS: a high capacity genotyping by sequencing analysis pipeline. PloS One 9:e90346
Goodstein DM, Shu S, Howson R et al (2012) Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res 40:1178–1186
Grady BJ, Torstenson ES, Ritchie MD (2011) The effects of linkage disequilibrium in large scale SNP datasets for MDR. BioData Min 4:11
He J, Zhao X, Laroche A, Lu ZX, Liu H, Li Z (2014) Genotyping-by-sequencing (GBS), an ultimate marker-assisted selection (MAS) tool to accelerate plant breeding. Front Plant Sci 5:484
Hill WG, Weir BS (1988) Variances and covariances of squared linkage disequilibria in finite populations. Theor Popul Biol 33:54–78
Kawano K (1980) Cassava. In: Fehr WR, Hadley HH (eds) Hybridisation of crop plants. America Society of Agronomy and Crop Science Society of America, Madison, pp 225–233
Korte A, Farlow A (2013) The advantages and limitations of trait analysis with GWAS: a review. Plant Methods 9:1–9
Li M, Liu X, Bradbury P et al (2014) Enrichment of statistical power for genome-wide association studies. BMC Biol 12:73
Lipka AE, Tian F, Wang Q et al (2012) GAPIT: genome association and prediction integrated tool. Bioinformatics 28:2397–2399
Manson J, Greiner T, Shrimpton R, Sanders D, Yukich J (2014) Vitamin A policies need rethinking. Int J Epidemiol. doi:10.1093/ije/dyu194
Mayer JE, Pfeiffer WH, Beyer P (2008) Biofortified crops to alleviate micronutrient malnutrition. Curr Opin Plant Biol 11:166–170
Monfreda C, Ramankutty N, Foley JA (2008) Farming the planet: geographic distribution of crop areas, yields, physiological types, and net primary production in the year 2000. Global Biogeochem Cycles 22:1–19
Montagnac AJ, Davis CR, Tanumuhardjo SA (2009) Nutritional value of cassava for use as a staple food and recent advances for improvement. Compr Rev Food Sci F 8:181–194
Moose SP, Mumm RH (2008) Molecular plant breeding as the foundation for 21st century crop improvement. Plant Physiol 147:969–977
Nassar NMA (2003) Cassava, Manihot esculenta Crantz genetic resources: anatomy of a diversity center. Genet Mol Res 2:214–222
Nassar NMA, Schwartz CA, Junior OP (2007) Cassava diversity in Brazil: the case of carotenoid-rich landraces. Genet Mol Res 6:116–121
Njoku DN, Vernon G, Egesi CN et al (2011) Breeding for enhanced ß-carotene content in cassava: constraints and accomplishments. J Crop Improv 25:560–571
Njoku DN, Gracen VE, Offei SK, Asante IK, Egesi CN, Kulakow P, Ceballos H (2015) Parent-offspring regression analysis for total carotenoids and some agronomic traits in cassava. Euphytica 206:657–666
Okogbenin E, Porto MCM, Egesi C, Mba C, Espinosa E, Santos LG (2007) Marker-assisted introgression of resistance to cassava mosaic disease into Latin American germplasm for the genetic improvement of cassava in Africa. Crop Sci 47:1895–1904
Oliveira EJ, Resende MDV, Silva Santos V et al (2012) Genome-wide selection in cassava. Euphytica 187:263–276
Owens BF, Lipka AE, Magallanes-Lundback M et al (2014) A foundation for provitamin A biofortification of maize: genome-wide association and genomic prediction models of carotenoid levels. Genetics 198:1699–1716
Paine JA, Shipton CA, Chaggar S et al (2005) Improving the nutritional value of golden rice through increased pro-vitamin A content. Nat Biotechnol 23:482–487
Pasam RK, Sharma R, Malosetti M, van Eeuwijk FA, Haseneyer G, Kilian B, Graner A (2012) Genome-wide association studies for agronomical traits in a world wide spring barley collection. BMC Plant Biol 12:16
Peterson G, Dong Y, Horbach C, Fu Y-B (2014) Genotyping-by-sequencing for plant genetic diversity analysis: a lab guide for SNP genotyping. Diversity 6:665–680
Pfeiffer WH, McClafferty B (2007) HarvestPlus: breeding crops for better nutrition. Crop Sci 47:88–105
Poland J, Endelman J, Dawson J et al (2012) Genomic selection in wheat breeding using genotyping-by-sequencing. Plant Genom J 5:103–113
Prochnik S, Marri PR, Desany B et al (2012) The cassava genome: current progress, future directions. Trop Plant Biol 5:88–94
R Development Core Team (2010) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Rabbi IY, Hamblin MT, Kumar PL, Gedil MA, Ikpan AS, Jannink JL, Kulakow PA (2014) High-resolution mapping of resistance to cassava mosaic geminiviruses in cassava using genotyping-by-sequencing and its implications for breeding. Virus Res 186:87–96
Remington D, Thornsberry JM, Matsuoka Y et al (2001) Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc Natl Acad Sci USA 98:11479–11484
Romay MC, Millard MJ, Glaubitz JC et al (2013) Comprehensive genotyping of the USA national maize inbred seed bank. Genome Biol 14:55
Salvador EM, Steenkamp V, McCrindle CME (2014) Production, consumption and nutritional value of cassava (Manihot esculenta Crantz) in Mozambique: an overview. J Agric Biotechnol Sustain Dev 6:29–38
Sánchez T, Chávez AL, Ceballos H, Rodriguez-Amaya DB, Nestel P, Ishitani M (2006) Reduction or delay of post-harvest physiological deterioration in cassava roots with higher carotenoid content. J Sci Food Agric 86:634–639
Semagn K, Bjørnstad A, Xu Y (2010) The genetic disection of quantitative traits in crops. Electro J Biotechn 13:1–45
Shumskaya M, Wurtzel ET (2013) The carotenoid biosynthetic pathway: thinking in all dimensions. Plant Sci 208:58–63
Shumskaya M, Bradbury LMT, Monaco RR, Wurtzel ET (2012) Plastid localization of the key carotenoid enzyme phytoene synthase is altered by isozyme, allelic variation and activity. Plant Cell 24:3725–3741
Stephenson K, Amthor R, Mallowa S et al (2010) Consuming cassava as a staple food places children 2–5 years old at risk for inadequate protein intake, an observational study in Kenya and Nigeria. Nutr J 9:9
Suwarno WB, Pixley KV, Palacios-Rojas N, Kaeppler SM, Babu R (2015) Genome-wide association analysis reveals new targets for carotenoid biofortification in maize. Theor Appl Genet 128:851–864
Swarts K, Li H, Romero Navarro JA et al (2014) Novel methods to optimize genotypic imputation for low-coverage, next-generation sequence data in crop plants. Plant Genome 7:1–12
Talsma EF, Melse-Boonstra A, de Kok BP, Mbera GN, Mwangi AM, Brouwer ID (2013) Biofortified cassava with pro-vitamin A is sensory and culturally acceptable for consumption by primary school children in Kenya. PloS One 8:e73433
Tan SL (2015) Cassava—silently, the tuber fills: the lowly cassava, regarded as a poor man’s crop, may help save the world from the curse of plastic pollution. Utar Agric Sci J 1:12–24
Turner SD (2014) qqman: an R package for visualizing GWAS results using Q-Q and Manhattan plots. http://dx.doi.org/10.1101/005165. Accessed on 12 June, 2014
Vazquez A, Bates D, Rosa G, Gianola D, Weigel K (2010) Technical note: an R package for fitting generalized linear mixed models in animal breeding. J Anim Sci 88:497–504
Wang M, Yan J, Zhao J, Song W, Zhang X, Xiao Y, Zheng Y (2012) Genome-wide association study (GWAS) of resistance to head smut in maize. Plant Sci 196:125–131
Weir BS (2008) Linkage disequilibrium and association mapping. Ann Rev Genomics Hum Genet 9:129–142
Welsch R, Arango J, Bar C et al (2010) Provitamin A accumulation in cassava (Manihot esculenta) roots driven by a single nucleotide polymorphism in a phytoene synthase gene. Plant Cell 22:3348–3356
WHO (2009) Global prevalence of vitamin A deficiency in populations at risk 1995–2005. WHO global database on Vitamin A deficiency. World Health Organisation, Geneva
Yuan-yuan L, Hong-jian T, Xiang-qian Z et al (2014) Molecular improvement of grain weight and yield in rice by using GW6 gene. Rice Sci 21:127–132
Acknowledgments
The authors are thankful to the East African Agricultural Productivity Project (EAAPP) for funding activities for field trials for this research. All the genotyping and bioinformatics analysis were done with support from the Buckler Laboratory at Cornell University.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
10681_2016_1772_MOESM4_ESM.tiff
Supplementary Fig. 2 Plot of genome-wide linkage disequilibrium decay based on adjacent pairwise genetic and physical distance. Pairs with distance >60 kb are not shown. Supplementary material 4 (TIFF 357 kb)
10681_2016_1772_MOESM5_ESM.tiff
Supplementary Fig. 3 Manhattan plots for genome-wide diagnosis of association signals based on means of total carotenoid content. P and Q are linkage groups containing unanchored scaffolds in the cassava genome sequence v6.1. Horizontal blue and red lines are the suggestive and 5 % Bonferroni threshold lines, respectively. Supplementary material 5 (TIFF 486 kb)
10681_2016_1772_MOESM6_ESM.tiff
Supplementary Fig. 4 Manhattan plot for genome-wide diagnosis of association signals based and root flesh colour. P and Q are linkage groups containing unanchored scaffolds in the cassava genome sequence v6.1. Horizontal blue and red lines are the suggestive and 5 % Bonferroni threshold lines, respectively. Supplementary material 6 (TIFF 461 kb)
Rights and permissions
About this article

Cite this article
Esuma, W., Herselman, L., Labuschagne, M.T. et al. Genome-wide association mapping of provitamin A carotenoid content in cassava. Euphytica 212, 97–110 (2016). https://doi.org/10.1007/s10681-016-1772-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10681-016-1772-5