
Abstract
Understanding the genetic basis underlying domestication-related traits (DRTs) of cowpea (Vigna unguiculata (L.) Walp.) is important since the genome has experienced divergent domestication and in addition it is also useful to utilize the wild germplasm efficiently for improving different traits of the cultivated cowpea. Quantitative trait loci (QTLs) for DRTs were identified in a population of 159 F7 recombinant inbred lines derived from a cross between a domesticated cowpea (V. unguiculata (L.) Walp.) variety, 524B, and a wild accession, 219. Using the constructed linkage map, QTLs for 10 DRTs were analysed and mapped. QTLs for seed, pod and flower related traits were detected. Subsequently, QTL for ovule number was also identified. To our knowledge, this is the first time a QTL for this trait has been observed. QTLs for DRTs show co-localization on three linkage groups and pleiotropy or close linkage of genes for the traits is suggested in these chromosome regions. The information gained in this study can be used for marker-assisted selection of domestication-related QTLs in cowpea and enhance understanding of domestication in the genus Vigna.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
According to Harlan (1992), cultivated plants were domesticated from their wild progenitors at about 10,000 years ago. Due to natural or deliberate human selection, the morphological and physiological changes associated with domestication can be delimitated into adaptation syndromes. Selection pressures that are associated with planting and harvesting have resulted in loss of pod shattering (PS) characteristics, fast and rapid germination through loss or reduction of germination inhibitors and increases in seedling vigor whereas human-selected changes have included more determinate plants, larger seeds, inflorescences by intentionally selecting for higher yield and other yield components (Gepts 2010). The genetic basis that underlies distinctive differences between cultivated crops and their wild relatives, known as the domestication syndrome has been investigated in several crop species such as rice (Xiong et al. 1999; Cai and Morishima 2002; Lee et al. 2005), maize (Doebley et al. 1990; Doebley and Stec 1993; Lauter and Doebley 2002), aubrgine (Doganlar et al. 2002), tomato (Grandillo and Tanksley 1996), pearl millet (Poncet et al. 2000, 2002), bean (Koinange et al. 1996), wheat (Peng et al. 2003), sunflower (Burke et al. 2002), soybean (Liu et al. 2007), azuki bean (Isemura et al. 2007; Kaga et al. 2008), rice bean (Isemura et al. 2010), cowpea (Andargie et al. 2011), yardlong bean (Kongjaimun et al. 2012), pigeonpea (Kumawat et al. 2012), mungbean (Isemura et al. 2012).
Ross-Ibarra (2005) reviewed the reports on quantitative trait locus (QTL) mapping of domestication-related traits (DRTs) and from most of these studies he was able to identify three major patterns. First, the QTLs found in apparently linked clusters in certain regions of the chromosome instead of randomly distributed throughout the genome. Secondly, relatively few genes or QTLs with large effects explain most of the variation associated with DRTs across a variety of taxa. Thirdly, there occurs synteny or these ‘domestication genes’ are conserved across species within a family, as in the case of the Gramineae (Paterson et al. 1995) and Solanaceae (Doganlar et al. 2002).
Domestication related traits as well as most of the agronomically important traits are quantitatively inherited and thus controlled by numerous genetic components (White and Doebley 1998). The development of easily usable and polymorphic DNA markers has facilitated QTL mapping and made it possible to understand the genetic architecture of quantitative traits including the number of loci underlying a trait, their chromosomal locations and their phenotypic effects (Tanksley 1993; Mackay 2001).
Cultivated and wild cowpeas belong to the same biological species and are constituents of the primary gene pool of Vigna. Vigna unguiculata (L.) Walp is composed of cultivated cowpea (V. unguiculata var unguiculata), subdivided in five cultigroups or varieties namely Unguiculata, Biflora, Melanophthalmus, Sesquipedalis and Textilis (Pasquet 1998). Wild gene pool includes Annual wild cowpea (V. unguiculata ssp. unguiculata var. spontanea) and ten wild perennial subspecies divided in two groups according to their breeding system. The perennial allo-autogamous subspecies (dekindtiana, stenophylla, tenius, alba and pubescens) are the most closely related to the annual forms than do the perennial allogamous subspecies (pawekiae, burundiensis, letouzeyi, baoulensis and aduensis) (Pasquet and Baudoin 2001). Crosses between them generally produce viable and fertile progeny (Kouadio et al. 2006; 2007). Cultivated and wild cowpeas differ in various morphological and physiological characteristics collectively designated as the domestication syndrome (Piper 1913; Faris 1965; Lush and Evans 1980).
The typical cultivated phenotype includes a determinate growth habit (GH), having large seeds with variable seed coat colors, a loss of seed dispersal ability and rapid & uniform germination while the wild phenotype is characterized by having an indeterminate GH, bearing small seeds. The wild cowpea also differs in the extent of hard seededness (HS) and pod dehiscence from the cultivated cowpea. The domestication syndrome is a suite of morphological and physiological traits that distinguish domesticated crops from their wild ancestors (Hammer 1984).
Crop domestication is an accelerated evolutionary process that is basically the result of the synergistic effect of human (both intentional and unintentional selection) and natural selection. Plant scientists are interested in studying the genetic basis of crop domestication with an ultimate goal of identifying useful allele(s), gene(s) or genomic region(s) that have not been exploited in the wild relatives of the cultivated crops and could have a positive impact on crop improvement. The search and localization for genes involved in crop domestication are performed by means of QTL analyses in many crops.
A considerable number of QTLs have been detected in marker-assisted studies for various quantitative characteristics including physiological and morphological traits in several crops. However, there are few studies that are carried out for DRTs in cowpea. Information on the genetics of DRTs would be useful for cowpea breeding programmes, and for comparative genome studies among members of the genus Vigna. The objective of the present study was to identify genomic regions controlling DRTs and to gain a better understanding of the genetic basis of morphological and physiological traits that differentiate cultivated and wild cowpeas by means of molecular mapping of recombinant inbred lines (RILs) resulting from a cross between a cultivated and wild cowpea.
Materials and methods
Mapping population
One-hundred and fifty nine (159) F7 RILs were developed by single-seed descent from a cross between 524B, a California black-eyed cowpea, and 219, a unique wild plant from subsp. unguiculata var. spontanea (Schweinf.) Pasquet (formerly subsp. dekindtiana sensu Verdc.). 524B was developed from a cross between California cultivars CB5 and CB3 and contains multiple resistance and agronomic characteristics, whereas 219 is the plant collected in the wild cowpea population from coastal Kenya (4°16′S 39°15′E, elevation 210 m, soil from Mariakani sandstone geological formation). The original 219 wild plant is still maintained vegetatively.
Phenotypic evaluation
A total of ten DRTs were evaluated (Table 1). Of these six were treated as quantitative whereas the remaining four were qualitative traits that differentiate cultivated and wild cowpea. The F7 population of 159 plants, together with the parents, were grown in the screen house at the International Center of insect physiology and ecology (ICIPE), Nairobi, Kenya from January to June, in 2010. For PS, mature but undehisced pods were sampled for each of the RILs and parental lines, and preserved as bulk at a room temperature for 1 week and scored visually as dehisced or not. After evaluation, seeds were threshed by hand, and used for scoring the 100 seed weight (SW) and HS. 100 SW was measured by weighing 100 seeds while HS was evaluated by two replications as a percentage of impermeable seeds out of 20 seeds after 24 h of immersion in 50 ml of distilled water in a 200 ml beaker. Pod fiber layer and seed coat thickness was measured in methylene blue stained cross sections of almost mature green pods and seed coats using a microscope equipped with an ocular micrometer respectively. Five different pods and seeds were measured and the figure used is an average of five measures. The pod color (PDC) was divided into three classes according to the degree of darkness (1, white; 2, light tan; and 3, black). Habit was evaluated by visual scoring (i.e. 0 for trailing and 1 for erect), and number of ovules were evaluated by counting the number of ovules per pod and by taking the average of five measures of pods. Data for days to flowering (DF) was collected by checking flowering daily and scored as the number of days from sowing to the first flower opening on each plant for all the 159 RILs and the two parents. Seeds of all the 159 F7 RILs and the two parents were surface sterilized and were sown in rock wool plug trays (25 × 25 × 40 mm/cell), which was moistened with water and placed in the growth chamber. To avoid heating of the pouches, the containers were covered with aluminum foil, leaving an opening 2 cm wide through which the seed grew. Fifteen to 21 days after sowing, the root system architecture (RSA) could be analyzed.
DNA extraction and polymerase chain reaction (PCR)
The DNA of the 159 F7 individuals and parental lines was extracted from 1 g of fresh leaf tissue according to a modified CTAB procedure (Mignouna et al. 1998). The cowpea leaf tissue was frozen in liquid nitrogen and then crushed in 1.5 ml microfuge tubes using a mini-pestle. Succeeding steps for polymerase chain reaction (PCR) were carried out following the methods described by Andargie et al. (2011). The PCR product was stored at 4 °C before analysis.
DNA gel electrophoresis
Amplified products were resolved using 6 % (19:1, acrylamide: bisacrylamide) non-denaturing polyacrylamide gel. About 2.5 μl of loading buffer (6×) consisting of 0.25 % bromophenol blue, 0.25 % xylene cyanol FF, 30 % glycerol and 5 mM EDTA (pH 8.0) was added to the PCR product and a 7 μl sample was loaded on the gel. Gels were run on 1 × TBE (90 mM Tris, 90 Mm boric acid, 2 mM EDTA and pH 8.3) buffer at 350 volts for 3.5 h in a Mega-gel High Throughput Vertical Unit (CBS Scientific, Del Mar, CA). Patterns were visualized under UV using Kodak molecular imaging systems (Eastman Kodak Company, Rochester, NY).
Linkage map construction and QTL analysis
Of the SSR markers tested, 202 polymorphic and informative SSR markers and 4 morphological markers were chosen to construct the linkage groups (LGs) covering all the 11 LGs. A total of 206 markers were mapped in the RIL population. SSR marker nomenclature was done by sequentially assigning numbers that are associated with particular gene space sequences from Cowpea Genespace Sequence Database. Each identified SSR gets a unique number and added to a list of known/confirmed SSRs. JoinMap 4.0 software was used for linkage analysis, map construction, and map drawing (Van Ooijen 2006). The Kosambi mapping function (Kosambi 1944) was used to convert recombination frequency to Centimorgans (cM).The map was constructed using a LOD value of 3.0 and a recombination frequency of 0.30 for all LGs. Marker segregation was subjected to the Chi square test to examine distortion from the expected 1:1 segregation.
All QTL analyses were performed using composite interval mapping (CIM; Zeng 1994) as implemented by the Qgene version 4.3 (Nelson 1997). This approach tests the hypothesis that an interval between two adjacent markers harbors a QTL affecting the trait of interest while controlling for the effects of other QTL segregating outside the region of interest. The LOD curves were created by scanning at 2 cM intervals, while a permutation test (1,000 resamplings) was performed to determine the critical LOD score appropriate to empirically identify a putative QTL with a genome-wide error at a 0.05 confidence level (Churchill and Doerge 1994). A minimum LOD score of 3.0 was used for the identification of putative QTLs, and the percentages of total phenotypic variation and additive effects explained by each QTL for the trait that we are interested in was also calculated. One-LOD support limits for the position of each QTL were calculated from the CIM results.
Results
Segregation of markers and map construction
Most of the markers used segregated in an expected 1:1 ratio in the RIL population. Twenty-two markers (10.67 % of the 206 markers) on 11 LGs produced a significantly (P ≤ 0.05) distorted segregation. Of these, the distortions observed in SSR6607, SSR6228 on LG 1 and SSR6348, SSR6451 on LG 2, were highly significant (P ≤ 0.01). Linkage map of 206 markers and covering 677 cM was constructed using the Kosambi function. Markers were randomly distributed on the 11 LGs with an interval between loci ranging from 0.5 to 22 cM. The 202 SSR and the four morphological markers were assigned to 11 LGs (Fig. 1) with 5–49 markers per LG and LG length varying from 11 cM (LG11) to 241 cM (LG1). As expected, SSR markers were evenly distributed and not clustered like amplified fragment length polymorphism (AFLP) markers (Menendez et al. 1997; Ouedraogo et al. 2002), which provide a better coverage of the genome of cowpea with markers uniformly distributed in each LG.

Genetic linkage map of cowpea harboring significant QTLs for DRTs in a RIL population derived from a cross between 524B and 219. DF days to flowering, SDP seed coat permeability, PFT pod fiber thickness, ON, number of ovules, SDG seed germination, SW seed weight
Segregation of domestication-related traits
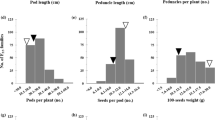
Significant differences were detected between the two parents for all the quantitative and qualitative traits listed in Table 1. The F7 segregation of the RILs for four qualitative traits; GH, PDC, root architecture and PS fit the expected 1:1 ratio. Frequency distributions of phenotypes in the RIL population were presented in Fig. 2. DTF, seed coat, seed germination (SDG) and pod fiber layer thickness (PFT) showed a bimodal distribution, suggesting an involvement of one or two major genetic factors. On the other hand, ovule number (ON), and SW were characterized by showing more-or-less normal distribution (Fig. 2), suggesting involvement of multiple genes. From the quantitative traits examined ON and DTF showed transgressive segregation (Fig. 2). The occurrence of this transgression could be linked to the presence of complementary QTL alleles in the two parental species and could also possibly suggest the existence of intragenic or intergenic interactions.

Histograms illustrating the frequency of DRTs for the 159 RILs of cowpea cross 524B × 219
Mapping of quantitative trait loci
The results of QTL analyses for each trait in the F7 population were shown in Table 2 and Fig. 1. In total, composite interval mapping revealed 15 major QTL for the six DRTs by the genome-wide analysis with permutation tests. Generally 1–7 QTL were detected for each trait at the level of significance (α = 0.05).
Seed germination (SDG)
One of the most important changes that occurred during the domestication of cowpea is selection for reduced seed dormancy and increased SDG after planting. For SDG, one QTL (qsdg1) was found on the LG 1 which showed a relatively high contribution (PEV 12.9 %). As expected, the allele from the cultivated parent on qsdg1 increased SDG. The cultivated-parent allele accelerated germination rate and was dominant over the wild-parent allele.
Seed coat permeability/seed coat thickness (SDP)
Seed coat permeability is one of the first steps in breaking dormancy and initiating germination in cowpea seed. For SDP, three QTL (qsdp2, qsdp10.1 and qsdp10.2) were detected on LG2 and LG10, respectively. qsdp2 on LG2 explained 12.2 % while qsdp10.1 and qsdp10.2 explained 10.3 and 9.9 % of the phenotypic variance respectively. The alleles of the wild parent additively decreased SDP at qsdp2 on LG2, while alleles from the cultivated parent dominantly increased seed-coat permeability at qsdp10 on LG10.
Pod fiber layer thickness (PFT)
Loss of pod dehiscence is advantageous for harvesting seeds while diminishing the chance for seed dispersal in natural habitats. One QTL, qpft5, with a relatively high contribution to this trait (10.8 %) was found at LG5. As expected, the allele from the wild parent decreased PFT. In addition, other four minor QTL (qpft1, qpft6 and qpft10) (LOD ≤ 2.7) with smaller effect were also found on LG1, LG6 (two QTL) and LG10.
Pod shattering (PS)
The QTL controlling PS was located at LG 5 between markers SSR6790 and SSR6623 and it was coincided with QTL position for PFT.
Seed weight (SW)
Domestication of cowpea has resulted in a sevenfold increase in SW (Table 1). For SW, seven QTL were mainly detected on LG1, LG2, LG3 (two QTL each on LG2 and LG3), LG7 and LG10. The alleles from the cultivated parent had a positive effect on the SW at LG 1 and at two QTL positions on LG 3 whereas alleles from the wild parent had a negative effect on this trait at LG7 and LG10 as well as at two QTL positions on LG 2. qsw1, qsw2.2, qsw3.2 and qsw7 on LG1, LG2, LG3 and LG7 revealed a relatively high contribution to this trait (19.1, 13.8, 13.3 and 15.6 %) while qsw3.1 and qsw10 on LG3 and LG10 explained 10.1 and 9.2 % of the phenotypic variance respectively.
Growth habit (GH)
The QTL controlling GH was mapped between markers SSR7079 and SSR7068 on LG1, and its position coincided with QTL positions for SW.
Root system architecture (RSA)
The QTL controlling RSA was mapped between markers SSR6609 and SSR6965-2 on LG1, and its position coincided with QTL positions for seed dormancy.
Days to flowering (DF)
Cultivated cowpea has a long pod filling period and thus flowers earlier than wild cowpea. The first flowering of the cultivated parent occurred earlier than the wild parent, 55 days compared with 152 days (Table 1). On the basis of the frequency distribution in F7 plants, the trait days to first flowering is controlled by a few genes. QTL analysis for DF revealed three QTLs. The QTL with the highest LOD value (qdf1) was located in LG1, and explained 18.5 % of the phenotypic variance (Table 2; Fig. 1). The recessive allele from the wild parent hastened flowering time mode. On the other hand, other two minor QTL (qdf2 and qdf7) (LOD ≤ 2.7) with smaller effect were found on LG2 and LG7. These alleles of the wild parent hastened flowering at QTL on LG2 and LG7.
Pod color (PDC)
The PDC of the cultivated parent is white whereas the wild parent is black. The QTL controlling PDC was mapped between markers SSR6302 and SSR6686 on LG2.
Ovule number (ON)
Two QTL (qon1 and qon3) related to the number of ovules detected and located on LGs 1 and 3. QTL detected on LG1 and LG3 explained 11 and 10.6 % of the phenotypic variance respectively. The alleles of the wild parent decreased ON at both qon1 and qon3 on LG 1 and 3 respectively. To the best of our knowledge this is the first report on QTLs that are regulating the ON in cowpea.
Distribution of domestication trait-related QTLs
Generally domestication related trait QTLs were distributed in six of the 11 LGs of cowpea. QTLs with large effect were found on LG1, LG2, LG3 and LG7 (Fig. 1) as described below.
LG1
QTLs for ON, DTF, SW and seed dormancy related traits were found on this region. QTL for ON (qon1) was found in this region near to a cowpea marker SSR7041 whereas QTL for DTF (qdtf1) was located near to a cowpea marker SSR6188. A QTL for seed dormancy (qsdg1) was also located in the same LG near to a cowpea marker SSR6243 whereas a QTL for SW (qsdw1) was located near to a cowpea marker SSR7117 on the same LG.
LG2
On this LG QTLs for SDP, SW and DTF were found. Between SSR markers SSR6360 and SSR6314, QTLs with a strong effect on DTF, SDP and SW were found.
LG3
QTL for SW (qsw3) and ON (qon3) with a relatively large phenotypic variance of 13.3 and 10.6 % was found in this region near to a cowpea marker SSR6924-2 and SSR6719 respectively.
LG7
This LG holds only one QTL which is responsible for SW. The QTL for SW (qsw7) was located near SSR marker SSR7027-2 and explains a phenotypic variance of 15.6 %.
Discussion
General features of the cowpea genetic linkage map
A total of 206 markers (202 SSR and four morphological markers) were included on this map. Markers were uniformly distributed in each LG and the interval between markers ranges from 0.5 cM to that of 22 cM. Therefore, this cowpea map is well suited for the identification of QTLs for DRTs and other agronomically important characters. It will also be useful for the analysis of genome synteny among species within the genus Vigna.
Segregation distortion
Segregation distortion of molecular markers has often been reported in the progeny resulting from crossing of cultivated crops × wild progenitors in maize (Doebley et al. 1990; Doebley and Stec 1993), rice (Cai and Morishima 2002), pearl millet (Poncet et al. 2000, 2002) and soybean (Keim et al. 1990; Yamanaka et al. 2001; Watanabe et al. 2004; Liu et al. 2007). Twenty-two markers (10.67 %) showed distorted segregation (P ≤ 0.05, Chi squared test) in the RIL population tested in this study. The results of linkage analysis revealed that markers with distorted segregation were distributed throughout the genome. Deviation from the expected segregation ratio was observed for markers on six LGs. The number of markers showing segregation distortion varied from one to eleven per LG. Segregation distortion of molecular markers may be associated with mapping population type, since a recombinant inbred population favors segregation distortion (Xu et al. 1997). In addition, during hybridization between genetically distant parents (e.g., cultivated and wild relatives), the alleles of wild species can be frequently lost, which leads to severe distorted segregation (Xu et al. 1997). However, here, the rather low percentage of distorted markers would suggest that 219 is not a parent too distant from domesticated cowpea.
Genomic regions and distributions of QTLs involved in cowpea domestication
Similar to the reports made in other crop plants like adzuki bean (Isemura et al. 2007; Kaga et al. 2008), common bean (Koinange et al. 1996), soybean (Liu et al. 2007), rice bean (Isemura et al. 2010), maize (Doebley et al. 1995) and rice (Xiong et al. 1999) in the present study also, it was revealed that DRTs in cowpea are found to be controlled by a few major genes together with some minor genes, and these are distributed within narrow regions on LGs. 15 QTLs for DRTs were distributed on six of the 11 LGs.
Seed coat permeability is one of the first steps in breaking dormancy and initiating germination in cowpea seed. In Vigna species, generally water enters the seed through the strophiole which is found near to the hilum. To determine the genetic factors related to physical dormancy in cowpea, the thickness of the seed testa and water absorption by the seeds was evaluated and four QTLs were identified on three LGs (three QTLs for thickness of the seed testa and one QTL for water absorption by the seeds respectively). Out of these QTLs, qsdp2 on LG2 for thickness of the seed coat and qsdg1 had the largest effect (12.9 %) each. Therefore, the structure of the strophiole may be controlled by one or more genes in this QTL region. Isemura et al. (2007), Kaga et al. (2008) reported two QTLs for SDWA in azuki bean and while Isemura et al. (2010) reported five QTLs in rice bean. In the present study, a linkage between QTLs for 100-seed weight (SDW) and thickness of the seed coat (SDP) was found on LG 10. So, it is necessary to elucidate whether the effects of these QTLs were caused by pleiotropic effects of the same gene or by distinct genes that were tightly linked. If the effects are proved to be caused by tight linkage rather than pleiotropy, it may be possible to separate the traits using marker assisted selection.
Generally, reducing the frequency of pod dehiscence is an important objective in plant breeding. PS is related to the strength and thickness of the pod fiber layer (Lush and Evans 1981). The trait is highly dependent on humidity. During very humid conditions (heavy rainfall outside the greenhouse), the pods from the wild plant do not shatter. On the other hand during very dry and hot conditions (for example during February in Nairobi) all the progenies with a visually intermediate phenotype were shattering, so looking for the QTLs as well as genes that are responsible for PS by measuring the PFT is crucial. In this study, one QTL for PFT was identified on LG5. The wild allele at this locus may be useful for improving pod dehiscence of the cultivated cowpea.
In cowpea SW is one of the three most important yield components and large seeds usually command consumer preference. In this study, seven QTLs for SW were found on five LGs. Fatokun et al. (1992) identified two QTL regions for SW. Unfortunately, a comparison of QTL regions between this study and that of Fatokun et al. (1992) was not possible because the kinds of markers used to construct the linkage map were different. Both parental types contributed alleles with positive effects on SW, which indicates that individuals with a potential for producing progenies with desirable allelic combinations for this trait could be identified in this cross. As it was observed in this study, alleles that improve the trait derived from parents that are agronomically inferior have been identified for several plant species (Swamy and Sarla 2008).
The QTL controlling GH was mapped between markers SSR7079 and SSR7068 on LG1, and its position coincided with QTL positions for SW. Similarly, Koinange et al. (1996) reported a single gene regulating a GH more specifically twining habit in common bean was located on chromosome one, this possibly suggest that similar mutations (genes) control determinacy in cowpea and common bean.
Root system architecture (RSA) describes the spatial organization of root systems, which is critical for root function in challenging environments (de Dorlodot et al. 2007). The root system provides, among others, anchoring and water/nutrient absorption to the plant. The architecture of a root system is the result of developmental processes and is a dynamic notion. The QTL controlling root architecture (rsa) was mapped between markers SSR6609 and SSR6965-2 on LG1, and its position coincided with QTL positions for seed dormancy.
Control of flowering time is a major target in plant breeding to produce novel varieties that better adapted to a particular environment (Jung and Muller 2009). Floral transition has been extensively studied in crop and model species and, in recent years, a growing number of related genes and QTLs have been identified (Kumar et al. 2011). In recent studies by Kongjaimun et al. (2012), Kumawat et al. (2012), 10 and 2 QTLs were identified for DF in yard long bean and pigeonpea, respectively. Here, we found three regions associated with DF. Large numbers of QTL below a certain level of statistical significance are usually ignored when determining the presence of a QTL (Churchill and Doerge 1994) so we reported here the major QTL that has a prominent effect. The two lines that were used as parents of the mapping populations differed for DF. DF was normally distributed, indicating a quantitative genetic control mechanism. Segregants were observed that flowered significantly earlier than 219 and later than 524B. One of the major causes of transgressive segregation in plants is the creation of novel combinations of complementary alleles from two parents, resulting in progeny with extreme phenotypes (Rick et al. 1976; De Vicente and Tanksley 1993). Although a major QTL, qdf1, was operating for the difference of DTF between 524B and 219, there were indications that other QTLs with relatively small effects were also operating in this cross.
The pattern of inheritance of PDC in cowpea is of great interest to breeders because its understanding could lead to the development of novel plant types with desirable PDC characteristics. PDC was controlled by a single QTL. The QTL controlling PDC was mapped between markers SSR6302 and SSR6686 on LG2. It was previously reported that the PDC in cowpea was controlled by a single recessive gene (Sangwan and Lodhi 1998).
Seed set is determined by the number of ovules per pod, the frequency of embryo abortions, and the number of pods per plant. Two relatively larger effect QTLs for ON (qon1-qon3) were found on LG1 and LG3 respectively, and the alleles from the wild parent increased these trait. Manipulation of these QTLs could be used to increase yield and seed number per pod in cowpea breeding. Fruit and seed development have been especially well-studied in soybean (Kato 1964; Shibles et al. 1975). Soybean ovaries contain anywhere from one to four ovules and the average number per pod is genetically determined (Shibles et al. 1975). The QTLs identified in this study for ON in cowpea were the first of its kind and they could possibly represent attractive targets for future cowpea breeding applications particularly for increasing of seed yield.
Co-localization of major QTLs
The QTLs controlling domestication traits were generally not randomly distributed across the crop genome (Gepts 2004). The distribution of domestication-related QTLs across the cowpea genome was shown by co-localization of the QTLs on different genomic regions and mainly on LG1, LG2 and LG10. QTLs for ON, seed dormancy, SW and DTF were co-located on LG1 whereas LG2 was associated with SW, DTF and seed coat thickness. In addition, the QTLs for seed coat thickness were co-localized with SW QTLs on LG10. QTL clusters for domestication were reported in several crops. The co-localization of QTL in part, may reflect non-random distribution of genes or it may suggest pleiotropy or both. Koinange et al. (1996) reported the determinacy gene (fin) has pleiotropic effects on the number of nodes on the main stem, the number of pods, and the number of DTF and maturity in common bean. In sunflower, the unbranched allele (B) decreased seed oil content and increased capitula diameter and SW (Bachlava et al. 2010).
Conclusions
A genetic linkage map of cowpea has been constructed using 206 SSR and morphological markers. In total, 17 QTLs for ten DRTs were identified, and QTLs with relatively large effects were distributed on seven out of the 11 LGs. In addition, cultivated cowpea has accumulated a considerable number of QTLs with small effects (PVE, <10 %). As a result, the accumulation of mutations with both large and small effects may have led to the differentiation between wild and cultivated cowpea. These results could be useful for the improvement of cowpea and related legumes. In addition, the results would be useful to introduce such unutilized genetic characteristics in the wild cowpea into the cultivated genetic background by marker assisted selection.
References
Andargie M, Pasquet RS, Gowda BS, Muluvi GM, Timko MP (2011) Construction of a SSR-based genetic map and identification of QTL for domestication traits using recombinant inbred lines from a cross between wild and cultivated cowpea (V. unguiculata (L.) Walp.). Mol Breed 28(3):413–420
Bachlava E, Tang S, Pizarro G et al (2010) Pleiotropy of the branching locus (B) masks linked and unlinked quantitative trait loci affecting seed traits in sunflower. Theor Appl Genet 120:829–842
Burke JM, Tang S, Knapp SJ, Rieseberg LH (2002) Genetic analysis of sunflower domestication. Genetics 161:1257–1267
Cai HW, Morishima H (2002) QTL clusters reflect character associations in wild and cultivated rice. Theor Appl Genet 104:1217–1228
Churchill GA, Doerge RW (1994) Empirical threshold values for quantitative trait mapping. Genetics 138:963–971
De Dorlodot S, Forster B, Page`s L, Price A, Tuberosa R, Draye X (2007) Root system architecture: opportunities and constraints for genetic improvement of crops. Trends Plant Sci 12:474–481
De Vicente MC, Tanksley SD (1993) QTL analysis of trans-gressive segregation in an interspeci®c tomato cross. Genetics 134:585–596
Doebley J, Stec A (1993) Inheritance of morphological differences between maize and teosinte: comparison of results for two F2 populations. Genetics 134:559–570
Doebley J, Stec A, Wendel J, Edwards M (1990) Genetic and morphological analysis of a maize teosinte F2 population—implications for the origin of maize. Proc Natl Acad Sci USA 87:9888–9892
Doebley J, Stec A, Gustus C (1995) Teosinte branched1 and the origin of maize: evidence for epistasis and the evolution of dominance. Genetics 141:333–346
Doganlar S, Frary A, Daunay MC, Lester RN, Tanksley SD (2002) Conservation of gene function in the Solanaceae as revealed by comparative mapping of domestication traits in eggplant. Genetics 161:1713–1726
Faris DG (1965) The origin and evolution of the cultivated forms of Vigna sinensis. Can J Genet Cytol 7:433–452. doi:10.1139/g65-058
Fatokun CA, Menancio-Hautea DI, Danesh D, Young ND (1992) Evidence for orthologus seed weight genes in cowpea and mung bean based on RFLP mapping. Genetics 132:841–846
Gepts P (2004) Crop domestication as a long time selection experiment. Plant Breed Rev 24:1–44
Gepts P (2010) Crop domestication as a long-term selection experiment. In: Janick J (ed) Plant breeding reviews: long-term selection: crops, animals, and bacteria, volume 24, part 2. John Wiley & Sons, Inc., Oxford. doi:10.1002/9780470650288.ch1
Grandillo S, Tanksley SD (1996) Analysis of horticultural traits differentiating the cultivated tomato from the closely related species Lycopersicon pimpinellifolium. Theor Appl Genet 92:935–951
Hammer K (1984) Das domestikationssyndrom. Kulturpflanze 32:11–34
Harlan JR (1992) Crops and man, 2nd edn. Am. Soc. Agronomy, Madison
Isemura T, Kaga A, Konishi S et al (2007) Genome dissection of traits related to domestication in azuki bean (Vigna angularis) and their comparison with other warm season legumes. Ann Bot 100:1053–1071
Isemura T, Kaga A, Tomooka N, Shimizu T, Vaughan DA (2010) The genetics of domestication of rice bean, Vigna umbellata. Ann Bot 106:927–944
Isemura T, Kaga A, Tabata S, Somta P et al (2012) Construction of a genetic linkage map and genetic analysis of domestication related traits in mungbean (Vigna radiata). PLoS ONE 7(8):e41304. doi:10.1371/journal.pone.0041304
Jung C, Muller AE (2009) Flowering time control and applications in plant breeding. Trends Plant Sci 14:563–573
Kaga A, Isemura T, Tomooka N, Vaughan DA (2008) The genetics of domestication of the adzuki bean (Vigna angularis). Genetics 178:1013–1036
Kato I (1964) Histological and embryological studies on fallen flowers, pods and abortive seeds in soybean, Glycine max (L.). Tokai-Kinki Natl Agric Exp Stn Bull 11:1–52
Keim P, Diers BW, Olson TC, Shoemaker RC (1990) RFLP mapping in soybean: association between marker loci and variation in quantitative traits. Genetics 126:735–742
Koinange EMK, Singh SP, Gepts P (1996) Genetic control of the domestication syndrome in common bean. Crop Sci 36:1037–1045
Kongjaimun A, Kaga A, Tomooka N et al (2012) An SSR-based linkage map of yardlong bean (Vigna unguiculata (L.) Walp. subsp. unguiculata Sesquipedalis Group) and QTL analysis of pod length. Genome 55:81–92
Kosambi DD (1944) The estimation of map distances from recombination values. Ann Eugen 12:172–175
Kouadio D, Toussaint A, Pasquet RS, Baudoin JP (2006) Barrières d’incompatibilité pré-zygotiques chez les hybrides entre formes sauvages du niébé, Vigna unguiculata (L.) Walp. Biotechnol Agron Soc Environ 10(1):1–9
Kouadio D, Echikh N, Toussaint A, Pasquet RS, Baudoin JP (2007) Organisation du pool génique de Vigna unguiculata (L.) Walp.: croisements entre les formes sauvages et cultivées du niébé. Biotechnol Agron Soc Environ 11:47–57
Kumar J, Choudhari AK, Solanki RK, Pratap A (2011) Towards marker-assisted selection in pulses: a review. Plant Breed 130:297–313
Kumawat G, Raje RS, Bhutani S et al (2012) Molecular mapping of QTLs for plant type and earliness traits in pigeonpea (Cajanus cajan L. Millsp.). BMC Genet 13:84–100
Lauter N, Doebley J (2002) Genetic variation for phenotypically invariant traits detected in teosinte: implications for the evolution of novel forms. Genetics 160:333–342
Lee SJ, Oh CS et al (2005) Identification of QTLs for domestication-related and agronomic traits in an Oryza sativa × O. rufipogon BC1F7 population. Plant Breeding 124:209–219
Lin Z, Griffith ME, Li X, Zhu Z, Tan L, Fu Y, Zhang W, Wang X, Xie D, Sun C (2007) Origin of seed shattering in rice (Oryza sativa L.). Planta 226:11–20
Liu Q, Ni Z, Peng H, Song W, Liu Z, Sun Q (2007) Molecular mapping of a dominant non-glaucousness gene from synthetic hexaploid wheat (Triticum aestivum L.). Euphytica 155:71–78
Lush WM, Evans LT (1980) The seed coats of cowpeas and other grain legumes: structure in relation to function. Field Crop Res 3:267–286
Lush WM, Evans LT (1981) The domestication and improvement of cowpeas. Vigna unguiculata (L.) Walp. Euphytica 30:579–587
Mackay TFC (2001) Quantitative trait loci in Drosophila. Nat Rev Genet 2:11–20
Menendez CM, Hall AE, Gepts P (1997) A genetic linkage map of cowpea (Vigna unguiculata) developed from a cross between two inbred, domesticated lines. Theor Appl Genet 95:1210–1217
Mignouna HD, Ellis NTH, Knox MR, Asiedu R, Ng QN (1998) Analysis of genetic diversity in Guinea yams (Dioscorea spp.) using AFLP fingerprinting. Trop Agric 75:224–229
Nelson JC (1997) QGENE: software for marker-based genomic analysis and breeding. Mol Breed 3:239–245
Ouedraogo JT, Gowda BS, Jean M, Close TJ, Ehlers JD, Hall AE, Gillaspie AG, Roberts PA, Ismail AM, Bruening G, Gepts P, Timko P, Belzile F (2002) An improved genetic linkage map for cowpea (Vigna unguiculata L.) combining AFLP, RFLP, RAPD, biochemical markers, and biological resistance traits. Genome 45:175–188
Pasquet RS (1998) Morphological study of cultivated cowpea Vigna unguiculata (L.) Walp. Importance of ovule number and definition of cv. gr Melanophthalmus. Agronomie 18:61–70
Pasquet RS, Baudoin JP (2001) Cowpea. In: Charrier A, Jacquot M, Hamon S, Nicolas D (eds) Tropical plant breeding. Science publishers, Enfield, pp 177–198
Paterson A, Lin Y, Li Z, Schertz K, Doebley J, Pinson S, Liu S, Stansel J, Irvine J (1995) Convergent domestication of cereal crops by independent mutations at corresponding genetic loci. Science 269:1714–1718
Peng J, Ronin Y, Fahima T, Ro¨der M, Li Y, Nevo E, Korol A (2003) Domestication quantitative trait loci in Triticum dicoccoides, the progenitor of wheat. Proc Natl Acad Sci USA 100:2489–2495
Piper CV (1913) The wild prototype of cowpea. USDA Bureau of Plant Industry. Circular No 124. Miscellaneous papers. WashingNeiton, Government Printing Office, pp 29–32
Poncet V, Lamy F, Devos KM, Gale MD, Sarr A, Robert T (2000) Genetic control of domestication traits in pearl millet (Pennisetum glaucum L., Poaceae). Theor Appl Genet 100:147–159
Poncet V, Martel E, Allouis S, Devos M, Lamy F, Sarr A, Robert T (2002) Comparative analysis of QTLs affecting domestication traits between two domesticated wild pearl millet (Pennisetum glaucum L., Poaceae) crosses. Theor Appl Genet 104:965–975
Rick CM, Kesicki E, Fobes JF, Holle M (1976) Genetic and biosystematic studies on two new sibling species of Lycopersicon frominterandean Peru. Theor Appl Genet 47:55–68
Ross-Ibarra J (2005) Quantitative trait loci and the study of plant domestication. Genetica 123:197–204
Sangwan RS, Lodhi GP (1998) Inheritance of flower and pod colour in cowpea (Vigna unguiculata (L.) walp). Euphytica 102:191–193
Shibles RM, Anderson IC, Gibson AH (1975) Soybean. In: Evans LT (ed) Crop physiology. Cambridge University Press, London, pp 151–189
Swamy BPM, Sarla N (2008) Yield-enhancing quantitative trait loci (QTLs) from wild species. Biotechnol Adv 26:106–120
Tanksley SD (1993) Mapping polygenes. Annu Rev Genet 27:205–233
Van Ooijen JW (2006) Join Map® 4, software for the calculation of genetic linkage maps in experimental populations. Plant Research International B. V. and Kyazma B. V. Wageningen, The Netherlands
Watanabe S, Tajuddin T, Yamanaka N, Hayashi M, Harada K (2004) Analysis of QTLs for reproductive development and seed quality traits in soybean using recombinant inbred lines. Breed Sci 54:399–407
White S, Doebley J (1998) Of genes and genomes and the origin of maize. Trends Genet 14:327–332
Xiong LZ, Liu KD, Dai XK, Xu CG, Zhang Q (1999) Identification of genetic factors controlling domestication-related traits of rice using an F2 population of a cross between Oryza sativa and O. rufipogon. Theor Appl Genet 98:243–251
Xu Y, Zhu L, Xiao J, Huang N, McCouch SR (1997) Chromosomal regions associated with segregation distortion of molecular markers in F2, backcross, doubled haploid, and recombinant inbred populations in rice (Oryza sativa L.). Mol Gen Genet 253:535–545
Yamanaka N, Ninomiya S, Hoshi M, Tsubokura Y, Yano M, Nagamura Y, Sasaki T, Harada K (2001) An informative linkage map of soybean reveals QTLs for flowering time, leaf morphology and region of segregation distortion. DNA Res 8:61–72
Zeng ZB (1994) Precision mapping of quantitative trait loci. Genetics 136:1457–1468
Acknowledgments
The authors wish to thank the members of the Timko lab for their help in various aspects of this work and for helpful suggestions on this manuscript, and Dr. Theresa Fulton for suggestions on data analysis, as well as Beatrice Elesani from ICIPE Muhaka field station (Kenya) for her contribution to the building of the recombinant inbred family. These studies were supported by funds from the Kirkhouse Trust and the German Academic Exchange Program (DAAD). While the research reported here was funded by the Kirkhouse Trust, the design, execution, and interpretation of the research remains wholly the responsibility of the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Andargie, M., Pasquet, R.S., Gowda, B.S. et al. Molecular mapping of QTLs for domestication-related traits in cowpea (V. unguiculata (L.) Walp.). Euphytica 200, 401–412 (2014). https://doi.org/10.1007/s10681-014-1170-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10681-014-1170-9