
Abstract
Direct seeding is becoming more popular mainly due to its labor-saving nature. However, flooding during germination caused by unleveled fields and unpredicted heavy rain can prevent crop establishment. On the other hand, flooding just after sowing protects the seeds from rats and birds and is also a viable means of weed control. Thus, the development of varieties able to tolerate flooding during germination, referred to as anaerobic germination (AG), is essential. A study was conducted to identify QTLs associated with tolerance of flooding during germination from an F2:3 mapping population derived from the cross of IR64 and the tolerant aus landrace Nanhi. Phenotyping was performed by counting the rate of seedling survival of 300 lines under the stress. Selective genotyping was employed by genotyping the 48 most tolerant and 48 most susceptible lines using a 384-plex SNP Indica/Indica set on the Illumina BeadXpress Reader, resulting in 234 polymorphic markers for the study. A major QTL for AG derived from Nanhi, named qAG7, was detected on chromosome 7 with an LOD of 13.93 and 22.3 % of the phenotypic variance explained. A second QTL of smaller effect, qAG11, was also derived from Nanhi, while one QTL with an increased effect from IR64 was detected on chromosome 2 (qAG2.1). The QTLs detected in this study can be used to further elucidate the mechanisms underlying AG tolerance in rice, and can also be used in marker-assisted selection and QTL pyramiding to provide higher AG tolerance to enable improved crop establishment in direct-seeded systems.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The high cost of labor and water requirements associated with rice transplanting means that farmers of both rainfed and irrigated rice fields need to shift to a more viable means of crop establishment such as direct seeding. Direct seeding also offers the advantage of earlier crop maturity, thus leading to double or even triple rice cropping. However, the yield of direct-seeded rice under farmers’ field conditions tends to be lower than that of transplanted rice (Pandey and Velasco 2002). This is due to poor, uneven establishment and inadequate control of weeds that lead to poor performance, thereby necessitating higher input to control weeds. Other constraints associated with direct seeding are difficulties in controlling pests such as rats, birds and snails, and flooding that occurs in unleveled fields or during unexpected rain (Pandey and Velasco 2002) which limits the availability of oxygen needed for plant growth. Breeding rice varieties that can germinate and emerge under flooded conditions (i.e. can tolerate anaerobic conditions during germination) will help overcome these problems. Flooding the field just after broadcasting seeds is also an effective and practical means of controlling weeds. The availability of such varieties could promote widespread adoption of direct seeding in both rainfed and irrigated rice production areas.
Several mechanisms associated with rice germination and survival under limited oxygen have been investigated. For example, the coleoptiles of some rice varieties can elongate faster under low oxygen concentrations, thereby enabling the growing seedlings to reach the water surface and come in contact with air (Ella and Setter 1999). In addition, α-amylase was reported to be the key enzyme in rice starch degradation under limited oxygen concentration, providing energy to the growing seedlings (Hwang et al. 1999). Under limited O2 availability, alcohol fermentation takes place instead of mitochondrial respiration. In this pathway, there are increases in pyruvate decarboxylase and alcohol dehydrogenase activities in tolerant genotypes, indicating that plants are able to adjust to O2 stress through ethanol fermentation (Ismail et al. 2009, 2012). The hormone ethylene also promotes elongation of the mesocotyl and coleoptiles in rice seedlings (Satler and Kende 1985). During germination, ethylene produced by the growing embryo may further promote cell expansion by down-regulating the action of peroxidase, the enzyme involved in enhanced cell-wall rigidity (Ismail et al. 2009). Rapid cell expansion is needed for fast growth under low oxygen availability. Expansins could also be involved in regulating cell-wall growth and elongation, as they are known to regulate cell-wall expansion and extensibility, and are involved in rice coleoptile elongation under anoxia (Huang et al. 2000; Lasanthi-Kudahettige et al. 2007). Although these mechanisms are involved in the rice plant response to AG conditions, more research is needed to characterize the key mechanisms and to identify the genes that control AG tolerance.
Efforts in breeding for tolerance of anaerobic germination (AG) and increased seedling vigor started in the 1990s (Yamauchi and Winn 1996; Redoña and Mackill 1996; Biswas and Yamauchi 1997). However, progress over the years has been slowed by the limited knowledge on the genetics of the trait, the involvement of complex mechanisms and the lack of an effective screening method (Seshu et al. 1988). The use of marker-assisted selection (MAS) may significantly overcome these hindrances due to its efficiency and effectiveness (Septiningsih et al. 2009; Septiningsih et al. 2013a). The key to a successful MAS program is the identification and mapping of large-effect quantitative trait loci (QTLs) that control the trait of interest. QTL mapping for tolerance of AG in rice has begun to identify promising loci that promote an increased ability for germination under flooding (Jiang et al. 2004; Jiang et al. 2006; Angaji et al. 2010; Septiningsih et al. 2013b). Jiang et al. (2004) identified QTLs on chromosomes 1, 2 and 7 from DV85 and on chromosome 5 from Kinmaze with phenotypic variance of 10.5–19.6 %. In another study (Jiang et al. 2006), QTLs were also found on chromosome 5 (R 2 = 15.51 %) but the donor of tolerance was USSR5, a japonica variety. On the other hand, Angaji et al. (2010) identified several QTLs on chromosomes 1, 2, 3, 7, 8 and 9 from Khao Hlan On, a landrace from Myanmar. The QTL on chromosome 9 had the largest phenotypic variance (33.5 %). Recently, Septiningsih et al. (2013b) identified six QTLs on chromosomes 2, 5, 6 and 7 from the landrace Ma-Zhan Red. All these studies used restriction fragment length polymorphism or simple sequence repeat (SSR) markers to determine the QTLs associated with AG. However, genotyping a large number of samples using SSRs is laborious and time-consuming. The availability of high-quality rice genome sequence (Yu et al. 2002, International Rice Genome Sequence Project 2005) has led to the discovery of single nucleotide polymorphisms (SNPs). SNPs are the most abundant of all markers in the rice genome and have drawn considerable interest from researchers because of the availability of SNP genotyping platforms that are amenable to multiplexed sets of markers for different applications (Thomson et al. 2012). In this study, we report on the use of the Illumina GoldenGate 384-plex Indica–Indica SNP set on the BeadXpress platform for SNP genotyping to identify the positions of QTLs underlying AG tolerance in rice.
Materials and methods
Plant materials and mapping population development
Nanhi, an AG tolerant upland landrace from India (Angaji et al. 2010; Ismail et al. 2009), was used as the donor of AG tolerance and was crossed to IR64, a semi-dwarf indica lowland elite modern variety developed at IRRI, which is sensitive to flooding during germination. Based on the Illumina BeadXpress 384-plex SNP analysis when compared with representative accessions from the five subgroups of cultivated rice, Nanhi is categorized in the aus subgroup (unpublished data). The true F1 plant was confirmed using SSR markers then allowed to self-pollinate to produce the F2 population for genotyping and F2:3 populations for phenotyping. A total of 300 F2:3 families were used for the QTL study.
Screening for AG tolerance
The screening for AG tolerance followed the protocol of Septiningsih et al. (2013b). Thirty healthy seeds from each of the 300 lines were sown into seedling trays at about 1 cm soil depth, with 11 lines per tray including both parents as controls. The rows between lines and the outside borders were left empty. The arrangement of lines was based on an alpha plus randomization and was replicated twice. After sowing, the trays were submerged carefully in concrete tanks filled with 10 cm of water head above the soil surface in the trays; and was maintained at that depth for 21 days. Seedling survival rate was assessed 21 days after sowing.
Genotyping using SNPs
Total genomic DNA of IR64, Nanhi and the F2:3 progenies (48 most tolerant and 48 least tolerant lines) was extracted from 18 to 21-day-old seedlings using a modified CTAB technique based on the protocol of Murray and Thompson (1980). The final concentration of the DNA samples was normalized to 50 ng/μL. Genotyping was performed using the Illumina BeadXpress 384-plex SNP plates with the oligo pool assay (OPA) customized for Indica–Indica (Illumina OPA ID: GS0011861-OPA; RiceOPA2.1) (Thomson et al. 2012). PCR amplification and hybridization were performed using the GoldenGate genotyping assay for VeraCode manual protocol (Illumina, San Diego, CA, USA). The VeraCode 384-plex plate was scanned using the Illumina BeadXpress Reader (Genotyping Services Lab, IRRI) and raw intensity values were exported from the GenomeStudio software V1.1.0 (Illumina) and genotype calling was performed using Alchemy software (Wright et al. 2010).
Linkage map construction and segregation distortion
The linkage map was constructed using Map Manager QTX, vQTXb20, with Kosambi mapping function (Manly et al. 2001). The observed segregation of each SNP marker was tested against the expected Mendelian ratio (1AA:2AB:1BB). A χ 2 test was used to determine whether a marker was distorted (\(\chi_{p < 0.05}^{{^{ 2} }} = 6.00;\chi_{p < 0.01}^{{^{ 2} }} = 9. 2 1\) ) using QTL Cartographer v2.5 (Wang et al. 2010). We inferred the positions of the centromeres along the chromosomes based on the rice physical map published by the International Rice Genome Sequencing Project (IRGSP) (2005).
QTL analysis
QTL analysis was performed using interval mapping (IM) and composite interval mapping (CIM) of QTL Cartographer v2.5 (Wang et al. 2010) with 1,000 permutations. The cofactors in CIM were selected automatically using forward–backward stepwise regression with F-in = 0.01 and F-out = 0.01 and window size of 10 cM (Septiningsih et al. 2012). The LOD values at p ≤ 0.05 for the permutations were used as a threshold to declare the significance of the QTLs. The data were also analyzed using QTLNetwork v2.1 (Yang and Zhu 2005; Yang et al. 2007; Yang et al. 2008), and digenic interactions were also tested. Permutation of 1,000 iterations was used to determine the threshold of the F-value of the QTL. The naming of the QTLs was in accordance with that proposed by McCouch and Committee on Gene Symbolization, Nomenclature and Linkage, Rice Genetics Cooperative (CGSNL) (2008). In order to determine whether the QTLs in this study are novel or the same as those previously reported, we compared the relative positions of the confidence intervals and QTL peaks.
Results
AG tolerance in an IR64/Nanhi population
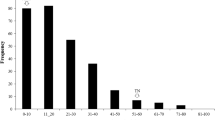
Nanhi, a tolerant upland landrace from India (Angaji et al. 2010; Ismail et al. 2009), served as the donor of AG tolerance and was crossed to IR64, a semi-dwarf indica lowland elite variety developed at IRRI, which is sensitive to flooding during germination. An IR64/Nanhi population of 300 F2:3 families showed an approximately normal distribution of survival under 10 cm water (skewness = −0.39 and kurtosis = −0.24), suggesting that AG tolerance is a quantitative trait controlled by multiple genes (Fig. 1). The survival rate differed greatly across the 300 lines, ranging from 3 to 90 %, with a mean of 48.7 %, while the survival rates of the parents were 23.3 % (IR64) and 59.5 % (Nanhi). Of the 300 lines, 8 % (24 lines) had a survival rate lower than IR64 while 36.7 % (110 lines) were higher than Nanhi. The data suggest that transgressive segregation exists in the population.

Phenotypic distribution of tolerance of anaerobic germination (AG) in an F2:3 IR64/Nanhi population. Evaluation of the survival rate (% survival) of the F2:3 mapping population was conducted under flooding conditions during germination. This figure shows the phenotypic distribution of tolerance of the 300 families. The average survival of the parents (IR64 and Nanhi) is also indicated
SNP-based linkage map
A total of 384 SNP markers were tested for polymorphism and, out of these, 234 (61 %) were found to be polymorphic between IR64 and Nanhi. These markers were used to genotype the 48 most tolerant and 48 least tolerant F2:3 families for a selective genotyping approach (i.e. selecting the two extreme tails of the phenotypic distribution) which decreases the cost of genotyping without compromising the power of detecting QTLs (Darvasi and Soller 1992). The rice physical map (MSU v.6) was used to order the markers for the SNP-based map, while the marker distances were calculated from the genotype data using MapManager QTX vQTXb20 (Manly et al. 2001). The total length of the map was 1564.1 cM, with an average of 7 cM distance between markers, which provides a well-covered map for QTL identification (Fig. 2).

Linkage map of SNP markers in IR64/Nanhi F2 population showing the positions of the QTLs. The SNP-based map of an F2:3 mapping population of IR64 × Nanhi was constructed using 234 markers. The figure shows the positions of the QTLs: square with bar indicates QTLs detected (p ≤ 0.05) in QTLNetwork; circle with bar indicates QTLs detected in QTL Cartographer (filled circles with continuous line bars are significant at p ≤ 0.05 while filled circles with broken line bars are QTLs just below the threshold level based on 1,000 permutations)
No recombination occurred between closely linked markers such as id2015762 and id20157627 on chromosome 2, id3200001 and id3008333 on chromosome 3 and id11003281 and id11003556 on chromosome 11; thus, the positions of those markers on each of those chromosomes were the same in cM distance. On the other hand, some sets of markers that were positioned in regions where recombination is limited (such as near or at the centromere) resulted in close genetic distance (cM), although they appear to be far in physical distance (Mb). Examples are regions on chromosomes 3 (22.41–23.02 Mb), 5 (9.23–13.73 Mb), 6 (14.6–17.5 Mb), 9 (1.3–4.2 Mb) and 11 (12.0–14.9 Mb) (Fig. 2).
Some segregation distortion was detected in the population: out of the 234 markers, 36 (15 %) showed over-representation of one of the genotype classes at p ≤ 0.05 and 22 of these showed highly significant (p ≤ 0.01) deviation. Most of the distorted markers occurred in regions on chromosomes 3, 4, 7 and 11. Thirty-four of the markers that showed distortion were skewed toward BB (Nanhi). Chromosomes 1 and 9 had only one marker each that slightly over-represented the heterozygous genotype (AB) and AA (IR64), respectively, while a region of distortion on chromosome 3 involved 18 markers from 97.7 to 188.8 cM (Suppl. Fig. 1). The marker id3011383 on chromosome 3 showed the highest distortion (X 2 = 56.25). A distorted region in the middle of chromosome 7 involved four markers from 46.6 to 64.6 cM, but did not exceed the 1 % significance threshold (X 2 = 9.20). The region on chromosome 4 had four distorted markers covering a region of 12.5–34.6 cM, with the most distorted marker being id4005120 with X 2 = 16.02.
QTL analyses
Based on 1,000 permutations using QTL Cartographer, the declared threshold levels at p ≤ 0.05 were 2.47 and 2.55, respectively, for interval mapping (IM) and composite interval mapping (CIM), while at p ≤ 0.01 they were 3.26 for IM and 3.52 for CIM. Two QTLs were detected above the empirical significance threshold at p ≤ 0.05: one on chromosome 7 and the other on chromosome 11 (Fig. 2; Table 1). In both QTLs, the Nanhi allele contributed to increased tolerance of AG. The QTL on chromosome 7 (qAG7) was significant at p ≤ 0.01 for both IM (LOD = 11.16) and CIM (LOD = 13.93). At the peak SNP marker, id7000988, the homozygous IR64 individuals had an average of 24.8 % survival, while the heterozygous class was 58.0 % and the homozygous Nanhi individuals averaged 60.3 % survival, indicating a largely dominant effect for this QTL. On the other hand, the QTL on chromosome 11 (qAG11) was significant in IM only, but just above the threshold level (p ≤ 0.05). The phenotypic variance (R2) ranged from 14.1 to 18.3 %. We reported qAG2.2 and qAG3 on chromosomes 2 and 3, respectively, that had LOD scores just below the threshold value for the purpose of detailed examination of the genetic basis of AG and comparison with other QTLs detected in other studies. The Nanhi allele acted to increase the tolerance of AG in qAG2.2, however, the increase effect of qAG3 came from the IR64 allele. The positions of these QTLs were also indicated on the linkage map (Fig. 2).
Using QTLNetwork, the threshold level to declare a QTL was p ≤ 0.05, with an F value at 13.56. Two QTLs were detected above the threshold: one is on chromosome 2 (qAG2.1) (Fig. 2), which was derived from IR64, and the other one was qAG7 derived from Nanhi, which was also detected by QTL Cartographer, with additive values (A) of 12.89 (p < 0.0001 and F = 14.90) and 22.26 (p < 0 and F = 43.70), respectively. The QTL heritability of the largest QTL, qAG7, was 0.33. No QTL interactions were detected in this population using QTLNetwork. The heritability for the trait observed in the populations was 0.61 and the standard deviation observed between the replicates for the phenotype was 3.18.
Discussion
Rice varieties that are tolerant of anaerobic conditions during germination are important to improve seedling establishment, especially when there are puddles of water due to unexpected rain or poor drainage. These varieties can also be directly broadcast onto the soil or sown in rows just below the soil surface and then flooded with a certain depth of water so that the seeds are protected from birds, rats and other predators. Flooding the seeds after broadcasting also protects them from being washed away by rain or seed drying in the case of pre-germinated seeds during a period of no rainfall (Pandey and Velasco 2002). Flooding the field is also an effective means of controlling weeds in a more sustainable way.
The survival rates of the parents used in this study were 59.5 % for Nanhi and 23.3 % for IR64. Phenotypic performance of the mapping population showed some extreme phenotypes relative to the parents used. This indicates that transgressive segregation appeared in the population and implies that both parents possess positive genes for AG tolerance and are located at different loci, and their recombination could result in superior genotypes. Similar results were described in both interspecific (Xie et al. 2008; Septiningsih et al. 2003; Thomson et al. 2003) and intraspecific (Guo et al. 2004; Zhou et al. 2007; Septiningsih et al. 2012) crosses in rice.
Segregation distortion
Markers that show segregation distortion and are clustered in regions along the chromosomes are referred to as segregation distortion regions (Xian-Liang et al. 2007) or segregation distortion loci (SDL) (Wang et al. 2005). According to Xu et al. (1997), a chromosomal region is considered to be associated with segregation distortion if at least four closely linked markers are significantly associated with the effect in a particular population. Considering these criteria, three chromosomal regions on chromosomes 3, 4 and 7 showed segregation distortion. Of the 36 markers that showed distortion, 72 % were clustered in these three chromosomal regions. The detected QTLs were not likely to be affected by the distortion, as the qAG3 was located in a region with just moderate distortion (not the chromosome 3 region with highest distortion), while qAG7 was in a different region than the distortion observed on chromosome 7. Moreover, it was reported that they do not generally pose problems in locating QTLs or in constructing linkage maps (Xu 2008).
Segregation distortion may be due to the proximity of markers with sterility (S) genes and gametophyte (ga) loci that have an effect on pollen abortion and competition ability (Liedl and Anderson 1993). In rice, more than 40 ga and S genes were reported (Nagato et al. 1998; Iwata et al. 1964; Zhao et al. 2006; Garavito et al. 2010). In this study, the regions with segregation distortion overlapped with previously reported ga and S genes and SDL such as ga-2, ga-3, s-e-1, S-9 and ga-14 in chromosome 3; ga-6, ga-10, ga-12, s-c-2, s-e-2 in chromosome 4, ga-11, S-7 and S-20 in chromosome 7 (Xu et al. 1997; Zhao et al. 2006; Garavito et al. 2010).
QTLs for AG tolerance
Using the software QTL Cartographer and QTLNetwork, we identified three QTLs above the permutation threshold: one each on chromosomes 2 (qAG2.1), 7 (qAG7) and 11 (qAG11). The results from two QTL mapping packages with different underlying algorithms were used to ensure a comprehensive view of all QTLs in the study. QTL Cartographer uses composite interval mapping with a stepwise cofactor selection across individual markers, while QTLNetwork uses marker-pair selection of cofactors which reduces the problem of overfitting and helps find the best-fitting models (Yang et al. 2007). In addition, QTLNetwork uses more conservative cofactor selection with a genome-wise false positive rate control, resulting in more stringent criteria for declaring a significant QTL (Yang et al. 2007; Yang et al. 2008). QTLNetwork also offers the advantage of detecting the presence of epistasis, although no interactions were detected in this study.
There are already a number of reported QTL studies on AG tolerance in rice (Jiang et al. 2004; Jiang et al. 2006; Angaji et al. 2010; Septiningsih et al. 2013b). A comprehensive comparison of previously identified QTLs indicated that none of the QTLs detected in this study are directly co-located with previously reported AG QTLs, suggesting that the landrace Nanhi likely contains novel genetic variation for anaerobic germination. For example, the major QTL on chromosome 7 (qAG7) in this study appears to be located in a different region to that of a major QTL (qAG7.1) derived from Ma-Zhan Red, which spans the region between RM3583 and RM21427 (Septiningsih et al. 2013b). However, even though the peak regions between the chromosome 7 QTLs between the two studies are more than 3 Mb apart, we cannot rule out the possibility that qAG7 from Nanhi may be the same as qAG7.1 from Ma-Zhan Red, since the confidence intervals are partially overlapping. Additional fine-mapping of both QTLs would be needed for more conclusive evidence. Nonetheless, even in the case that the two QTLs are controlled by the same gene, it will be interesting to study the origin and evolution, considering that Ma-Zhan Red is an indica landrace from China whereas Nanhi is an aus landrace from India.
High seedling vigor plays a major role in crop establishment and early vegetative growth in rice that lead to good field emergence, good crop performance and higher yields under different conditions (Foolad et al. 2007). Likewise, seeds with strong vigor have high competitive ability against weeds, which are a common problem associated with direct-seeded rice. Moreover, healthy seedling growth under low temperature imparts stable seedling establishment (SES), especially in DSR. Tolerance of germination at low temperature is one important objective of temperate rice breeding programs. Interestingly, qAG7 from the current study is in a similar position as that of a previously reported QTL for stable seeding establishment, qSES7-1 positioned between RM7161 and RM3635 (Iwata et al. 2010). In addition, qAG2.2 in this study is in a similar position as two previously reported QTLs for cold tolerance at the seedling stage, including qCTS-2 that is flanked by RM561-RM341 (Lou et al. 2007) and qLVG2 for low-temperature vigor of germination QTL between RM29 and RM262 (Han et al. 2006).
In summary, our data suggests that the QTLs detected in this study are likely novel, including the largest QTL on chromosome 7. These QTLs provide valuable targets to characterize the mechanisms underlying anaerobic germination and to rapidly transfer tolerance of AG into susceptible but otherwise elite varieties using marker-assisted selection. Since our study provided QTL targets that explain a moderate proportion of the phenotypic effect, they may need to be pyramided with QTLs from other populations to provide higher tolerance. Furthermore, this trait can also be combined with other abiotic stress-tolerance genes/QTLs such as those controlling submergence (SUB1) and salinity (Saltol) to provide more resilient rice varieties for directed-seeded cultivation in unfavorable environments.
References
Angaji SA, Septiningsih EM, Mackill DJ, Ismail AM (2010) QTLs associated with tolerance of flooding during germination in rice (Oryza sativa L.). Euphytica 172:159–168
Biswas JK, Yamauchi M (1997) Mechanism of seedling establishment of direct-seeded rice (Oryza sativa L.) under lowland conditions. Bot Bull Acad Sin 38:29–32
Darvasi A, Soller M (1992) Selective genotyping for determination of linkage between a marker locus and a quantitative trait locus. Theor Appl Genet 85:353–359
Ella ES, Setter TL (1999) Importance of seed carbohydrates in rice seedling establishment under anoxia. Acta Hortic 504:209–216
Foolad MR, Subbiah P, Zhang L (2007) Common QTL affect the rate of tomato seed germination under different stress and nonstress conditions. Int J Plant Genomics. doi:10.1155/2007/97386
Garavito A, Guyot R, Lozano J, Gavory F, Samain S et al (2010) A genetic model for the female sterility barrier between Asian and African cultivated rice species. Genetics 185:1425–1440
Guo L, Zhu L, Xu Y, Zeng D, Wu P, Qia Q (2004) QTL analysis of seed dormancy in rice (Oryza sativa L.). Euphytica 140:155–162
Han LZ, Zhang YY, Qiao YL, Cao GL, Zhang SY, Kim JH, Koh HJ (2006) Genetic and QTL analysis for low-temperature vigor of germination in rice. Acta Genet Sin 33:998–1006
Huang J, Takano T, Akita S (2000) Expression of α-expansin genes in young seedlings of rice. Planta 211:467–473
Hwang YS, Thomas BR, Rodriguez RL (1999) Differential expression of rice α-amylase genes during seedling development under anoxia. Plant Mol Biol 40:911–920
International Rice Genome Sequencing Project (IRGSP) (2005) The map-based sequence of the rice genome. Nature 436:793–800
Ismail AM, Ella ES, Vergara GV, Mackill DJ (2009) Mechanisms associated with tolerance to flooding during germination and early seedling growth in rice (Oryza sativa L.). Ann Bot 103:197–209
Ismail AM, Johnson DE, Ella ES, Vergara GV, Baltazar AM (2012) Adaptation to flooding during emergence and seedling growth in rice and weeds, and implications for crop establishment. AoB Plants. doi:10.1093/aobpla/pls019
Iwata N, Nagamatsu J, Omura T (1964) Abnormal segregation of waxy and apiculus coloration by a gametophyte gene belonging to the first linkage group in rice. Jap J Breed 14:33–39
Iwata N, Shinada H, Kiuchi H, Sato T, Fujino K (2010) Mapping of QTLs controlling seedling establishment using a direct seeding method in rice. Breed Sci 60:353–360
Jiang L, Hou M, Wang C, Wan J (2004) Quantitative trait loci and epistatic analysis of seed anoxia germinability in rice (Oryza sativa L.). Rice Sci 11:238–244
Jiang L, Liu S, Hou M, Tang J, Chen L, Zhai H, Wan J (2006) Analysis of QTLs for seed low temperature germinability and anoxia germinability in rice (Oryza sativa L.). Field Crops Res 98:68–75
Lasanthi-Kudahettige R, Magneschi L, Loret E, Gonzali S, Licausi F, Novi G, Beretta O, Vituli F, Alpi A, Perata P (2007) Transcript profiling of the anoxic rice coleoptile. Plant Physiol 144:218–231
Liedl B, Anderson NO (1993) Reproductive barriers: identification, uses and circumvention. Plant Breed Rev 11:11–154
Lou Q, Chen L, Sun Z, Xing Y, Li J, Xu X et al (2007) A major QTL associated with cold tolerance at seedling stage in rice (Oryza sativa L.). Euphytica 158:87–94
Manly KF, Cudmore RH Jr, Meer JM (2001) Map Manager QTX, cross-platform software for genetic mapping. Mamm Genome 12:930–932
McCouch SR, Committee on Gene Symbolization, Nomenclature and Linkage, Rice Genetics Cooperative (CGSNL) (2008) Gene nomenclature system for rice. Rice 1:72–84
Murray MG, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucl Acids Res 8:4321–4325
Nagato Y, Yoshimura A, Conveners (1998) Report of the committee on gene symbolization, nomenclature and linkage groups. Rice Genetics Newsletter 15:34–74
Pandey S, Velasco L (2002) Economics of direct seeding in Asia: patterns of adoption and research priorities. In: Pandey S, Mortimer M, Wade L, Tuong TP, Lopez K, Hardy B (eds) Direct seeding: research issues and opportunities. Proceedings of the International Workshop on Direct Seeding in Asian Rice Systems: Strategic Research Issues and Opportunities, 25–28 Jan 2000, Bangkok, Los Baños. International Rice Research Institute, pp 3–14
Redoña ED, Mackill DJ (1996) Genetic variation for seedling-vigor traits in rice. Crop Sci 36:285–290
Satler SO, Kende H (1985) Ethylene and the growth of rice seedlings. Plant Physiol 79:194–198
Septiningsih EM, Prasetiyono J, Lubis E, Tai TH, Tjubaryat T, Moeljopawiro S, McCouch SR (2003) Identification of quantitative trait loci for yield and yield components in an advanced backcross population derived from the Oryza sativa variety IR64 and the wild relative O. rufipogon. Theor Appl Genet 107:1419–1432
Septiningsih EM, Pamplona AM, Sanchez DL, Neeraja CN, Vergara GV, Heuer S, Ismail AM, Mackill DJ (2009) Development of submergence tolerant rice cultivars: the Sub1 locus and beyond. Ann Bot 103:151–160
Septiningsih EM, Sanchez DL, Singh N, Sendon PMD, Pamplona AM, Heuer S, Mackill DJ (2012) Identifying novel QTLs for submergence tolerance in rice cultivars IR72 and Madabaru. Theor Appl Genet 124:867–874
Septiningsih EM, Collard BCY, Heuer S, Bailey-Serres J, Ismail AM, Mackill DJ (2013a) Applying genomics tools for breeding submergence tolerance in rice. In: Varshney RK, Tuberosa R (eds) Translational genomics for crop breeding, vol 2., Improvement for abiotic stress, quality and yield improvementWiley-Blackwell, Hoboken, pp 9–30
Septiningsih EM, Ignacio JCI, Sendon PMD, Sanchez DL, Ismail AM, Mackill DJ (2013b) QTL mapping and confirmation for tolerance of anaerobic conditions during germination derived from the rice landrace Ma-Zhan Red. Theor Appl Genet 126:1357–1366
Seshu DV, Krishnasamy V, Siddique SB (1988) Seed vigor in rice. Rice seed health. International Rice Research Institute, Manila, pp 315–329
Thomson MJ, Tai TH, McClung AM, Hinga ME, Lobos KB, Xu Y, Martinez C, McCouch SR (2003) Mapping quantitative trait loci for yield, yield components, and morphological traits in an advanced backcross population between Oryza rufipogon and the Oryza sativa cultivar Jefferson. Theor Appl Genet 107:479–493
Thomson MJ, Zhao K, Wright M, McNally KL, Rey, Tung CW, Reynolds A, Scheffler B, Eizenga G, McClung A, Kim H, Ismail AM, de Ocampo M, Mojica C, Reveche MY, Dilla-Ermita CJ, Mauleon R, Leung H, Bustamante C, McCouch SR (2012) High-throughput single nucleotide polymorphism genotyping for breeding applications in rice using the BeadXpress platform. Mol Breed 29:875–886
Wang C, Zhu C, Zhai H, Wan J (2005) Mapping segregation distortion loci and quantitative trait loci for spikelet sterility in rice (Oryza sativa L.). Genet Res 86:97–106
Wang S, Basten CJ, Zeng Z-B (2010) Windows QTL Cartographer 2.5. Department of Statistics, North Carolina State University, Raleigh. http://statgen.ncsu.edu/qtlcart/WQTLCart.htm. Accessed 20 Oct 2013
Wright MH, Tung CW, Zhao K, Reynolds A, McCouch SR, Bustamante CD (2010) ALCHEMY: a reliable method for automated SNP genotype calling for small batch sizes and highly homozygous populations. Bioinformatics 26:2952–2960
Xian-Liang S, Xue-Zhen S, Tian-Zhen Z (2007) Segregation distortion and its effect on genetic mapping in plants. Chin J Agric Biotechnol 3:163–169. doi:10.1079/CJB2006110
Xie XB, Jin FX, Song MH, Suh JP, Hwang HG, Kim YG, McCouch SR, Ahn SN (2008) Fine mapping of a yield-enhancing QTL cluster associated with transgressive variation in an Oryza sativa × O. rufipogon cross. Theor Appl Genet 116:613–622
Xu S (2008) Quantitative trait locus mapping can benefit from segregation distortion. Genetics 180:2201–2208
Xu Y, Zhu L, Xiao J, Huang N, McCouch SR (1997) Chromosomal regions associated with segregation distortion of molecular markers in F2, backcross, doubled haploid, and recombinant inbred populations in rice (Oryza sativa L.). Mol Gen Genet 253:535–545
Yamauchi M, Winn T (1996) Rice seed vigor and seedling establishment in anaerobic soil. Crop Sci 36:680–686
Yang J, Zhu J (2005) Methods for predicting superior genotypes under multiple environments based on QTL effects. Theor Appl Genet 110:1268–1274
Yang J, Zhu J, Williams RW (2007) Mapping the genetic architecture of complex traits in experimental populations. Bioinformatics 23:1527–1536
Yang J, Hu CC, Hu H, Yu RD, Xia Z, Ye XZ, Zhu J (2008) QTLNetwork: mapping and visualizing genetic architecture of complex traits in experimental populations. Bioinformatics 24:721–723
Yu J, Hu S, Wang J, Wong G, Li S, Liu B, Deng Y, Dai L, Zhou Y, Zhang X, Cao M, Liu J, Sun J, Tang J, Chen Y, Huang X, Lin W, Ye C (2002) A draft sequence of the rice genome. Science 296:79–92
Zhao B, Deng Q-M, Zhang Q-J, Li JQ, Ye SP, Liang YS, Peng Y, Li P (2006) Analysis of segregation distortion of molecular markers in F2 population of rice. Acta Genet Sinica 33:449–457
Zhou L, Wang J-K, Yi Q, Wang YZ, Zhu YG, Zhang ZH (2007) Quantitative trait loci for seedling vigor in rice under field conditions. Field Crops Res 100:294–301
Acknowledgments
We thank R. Garcia, J. Mendoza, J. A. Tarun, V. Bartolome and C. J. Dilla-Ermita for technical assistance, and B. Hardy for editing the manuscript. The work reported here was supported in part by a grant from the Bill & Melinda Gates Foundation (BMGF) through the project “Stress-Tolerant Rice for Africa and South Asia (STRASA)” and by the Global Rice Science Partnership (GRiSP).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Baltazar, M.D., Ignacio, J.C.I., Thomson, M.J. et al. QTL mapping for tolerance of anaerobic germination from IR64 and the aus landrace Nanhi using SNP genotyping. Euphytica 197, 251–260 (2014). https://doi.org/10.1007/s10681-014-1064-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10681-014-1064-x