
Abstract
Association mapping relies on the variation and extent of linkage disequilibrium within a species to identify genes of interest; this is an alternative to linkage mapping in traditional biparental population, which exploits only the variation in the two parents of the mapping population. This study was designed to identify association between 48 SSR markers and downy mildew (DM) resistance using a set of 60 public and private maize inbred lines in Thailand. Genetic diversity and population structure in the set were calculated. A total of 489 alleles with an average gene diversity of 0.70 revealed two subpopulations among the 60 maize inbred lines. Analysis of variance resulted in significant effects of phenotypic values of tested entries, with significant effects of inbred lines, locations, and their interaction. In addition, the analysis of variance for broad sense heritability also indicated high heritability (0.97), and association analysis revealed three significant SSR-trait associations (P < 0.05). These three significant SSR loci have not been reported in previous linkage mapping studies. Our results suggest that new allelic variants associated with DM resistance in these germplasm collections should be useful to help identify new lines carrying alleles for DM resistance in breeding marker-assisted selection programs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Maize is one of five major crops grown in the uplands of Thailand (Ekasingh et al. 2004). It is predominantly used for animal feed, with 80–100 % of production sold to commercial poultry and livestock feed mills. It is a highly commercial crop handled by an extensive network of merchants. Maize sold as animal feed is mainly used domestically, and only a small fraction is exported or used for food (mainly white corn or sweet corn). Downy mildew (DM), caused by several species in the genera Peronosclerospora, Sclerospora and Sclerophthora, is one of the most destructive maize diseases in subtropical and tropical regions of Asia, including Thailand where the main causal agent is Peronosclerospora sorghi (Weston and Uppal) C.G. Shaw (Sriwatanapongse et al. 1993; Jeffers et al. 2000; Raymundo 2000; George et al. 2003).
In tropical and subtropical lowland Asia, maize-growing areas report economic losses due to downy mildew (Jeffers et al. 2000), occasionally as high as 75 % (Exconde and Raymundo 1974). Genetic resistance is a cost-effective and environmentally safe method for controlling downy mildew. However, despite the use of downy mildew resistant cultivars and metalaxyl fungicide as a seed treatment, the incidence of the disease is still severe in localized areas (Dalmacio 2000). New sources of locally adapted resistant lines may alleviate this.
Studies of the genetic basis of downy mildew resistance have been complicated by the polygenic nature of the trait and by the fact that additive effects contribute to resistance (Borges 1987; De Leon et al. 1993; Kaneko and Aday 1980; Singburaudom and Renfro 1982). Linkage mapping has been used as a tool of choice for the identification of quantitative trait loci (QTLs) that confer resistance to maize downy mildew (Agrama et al. 1999; George et al. 2003; Nair et al. 2005; Sabry et al. 2006; Jampatong et al. 2008). QTLs from various genomic regions on chromosomes 1, 2, 6, 7 and 10 have been found to confer resistance to downy mildew (George et al. 2003). The QTL approach has some limitations, including high costs and poor resolution in defining QTLs. Furthermore, with bi-parental crosses of inbred lines, only two alleles can be studied at any given locus.
Recently, a set of cultivars, lines, or landraces have been used to identify marker-trait associations in plants. This method uses linkage disequilibrium (LD) between DNA polymorphisms and genes underlying agronomic traits of interest (Thornsberry et al. 2001; Flint-Garcia et al. 2005; Yu et al. 2006; Buckler and Gore 2007; Zhu et al. 2008). Gene mapping through association studies has several advantages in traditional biparental populations because they can precisely pinpoint the genomic region responsible for the expression of the target trait, and have the potential to evaluate a large number of alleles per locus (Buckler and Thornsberry 2002; Flint-Garcia et al. 2003; Flint-Garcia et al. 2005). Informative polymorphisms identified in gene haplotypes and closely associated with a contrasting phenotype can be converted into DNA markers for use in marker-assisted selection (MAS). The application of gene mapping though association to plant breeding appears to be a promising approach to overcome some of the limitations of conventional linkage mapping, and to complete it (Stich et al. 2005; Yu and Buckler 2006).
The objective of this research was to identify and integrate SSR markers for gene mapping of DM resistance in a set of 60 maize inbred lines though linkage and association.
Materials and methods
Maize inbred lines
This study consisted of a set of 60 maize inbred lines (Table 1) supplied by two public-sector institutions and two private companies in Thailand. The National Corn and Sorghum Research Center (NCSRC-IICRD KU; Suwan Farm) and Nakhon Sawan Field Crop Research Center (NFR) supplied 17 and 15 inbred lines of field corn, respectively. Bangkok Seeds Industry and Sweet Seeds Company provided 15 inbred lines (7 field corn, 4 sweet corn, 2 waxy corn, 2 popcorn), and 13 inbred lines (11 sweet corn, 2 waxy corn), respectively.
Statistical design
Field experiments were conducted at two locations: Nakhon Sawan Field Crop Research Center (NFR) (15°20′45″N, 100°29′4″E) and the National Corn and Sorghum Research Center, Inseechandrastitya Institute for Crop Research and Development (NCSRC-IICRD KU) (14°24′42″N, 101°25′18″E), Thailand. All maize inbred lines were laid out in a randomized complete block design with three replicates during the 2008 rainy season (May–July). Two-row plots of 5 m in length, and 0.75 m wide of row spacing, were used for planting with 42 plants per plot using hand jab planter.
Methods of downy mildew inoculation
The spreader-row technique consisted of planting a susceptible variety (Tuxpeno-1 Sel. Sequia C3) in dense stands every 20 rows and in the alleyways was used as artificial inoculation of downy mildew in the fields. Seven to 8 days after planting (the first full leaf stage of development), the seedlings were sprayed using backpack sprayer with a spore suspension of the pathogen. To obtain a spore suspension, infected leaves were collected, washed, and incubated in a dark room for 8 h at 20–23 °C to induce sporulation. Spores were then washed from the leaves and inoculated on the spreader rows. To increase humidity, sprinkle irrigation was applied to the spreader rows for 1–2 h on the evening of the inoculation date. Artificial inoculation with sufficient moisture on the spreader rows was performed for two nights to allow for proper infection. Thereafter, sprinkle irrigation was applied to the spreader rows for several subsequent days to create a favorable environment for inoculation to develop infection. After 3 weeks, when the sufficient symptoms of infection appeared on the spreader rows, the study materials were planted, and the symptoms of infection was assessed by stand-counting of plants, followed up with assessments of infection one and 2 weeks later. Percentage disease was calculated as the ratio of the number of systemically infected plants to the total number of plants multiplied by 100.
DNA isolation and SSR
Genomic DNA was isolated from young leaves and bulk sample from five seedlings per variety using the modified CTAB method (Doyle and Doyle 1990). SSR primers were chosen from MaizeGDB (www.maizegdb.org) and tested in a preliminary experiment (Table 2). A panel of 48 polymorphic SSR loci from all 10 maize chromosomes was ultimately selected for the analysis (Phumichai et al. 2008). The SSR amplification reactions were carried out in 96-well microtiter plates using a PTC-200 Thermal Cycler (MJ Research, Watertown, MA, USA). The touchdown PCR parameters (Don et al. 1991) consisted of a cycle of 1 min at 96 °C, 1 min at 65 °C, and 1 min 30 s at 72 °C. The annealing temperature 65 °C was then reduced by 1 °C each cycle until a final annealing temperature of 55 °C was reached. The last cycle was then repeated 30 times and terminated with a continuous incubation at 4 °C. The 20 ml reaction mixture consisted of 20 ng of each primer, 0.5 units of Taq DNA polymerase (Fermentas), 200 mM of each dNTP, 1.5 mM MgCl2, 1× reaction buffer (10 mM Tris–HCl, 50 mM KCl, 0.1 mg/ml gelatin; pH 8.3), 20 ng template DNA, and de-ionized RO water. After amplification, reactions were stopped with 50 ml of loading dye (95 % formamide, 0.25 % bromophenol blue and 0.25 % xylene cyanol), heated to 95 °C for 3 min and then immediately placed on ice. Three milliliters of the sample were separated by electrophoresis in 6 % denaturing polyacrylamide gels (Sequi-Gen1 GT Nucleic Acid Electrophoresis Cell, Bio-Rad) at 40 W constant powers for 2 h and visualized by silver staining as described by Benbouza et al. (2006). The band sizes were compared using a 10 bp DNA ladder (Invitrogen).
Statistical analysis
Analysis of variance was performed using the IRRISTAT (International Rice Research Institute, ver. 5.0, Philippines) Coefficients of broad-sense heritability (H2), (Fehr 1987) by the equation \( H^{2} = \sigma_{G} /(\sigma_{E} /RL + \sigma_{GLe} /L + \sigma^{2} _{G} ) \) where σ 2 G , σ 2 E and σ 2 GL are genotypic variance, experimental error variance, and genotype × environment variance, respectively, and L and R are the number of locations and the number of replicates, respectively.
Population structure and genotypic data analyses
Gene diversity (GD), allele number, polymorphic information content (PIC) and Nei’s genetic distance were measured by calculating shared allele frequencies (Weir 1996) using PowerMarker 3.25 (Liu and Muse 2005). The UPGMA algorithm was used to construct a neighbor-joining dendrogram from a distance matrix based on Nei’s (1972) genetic distance, using MEGA4 software (Tamura et al. 2007) implemented in PowerMarker 3.25 (Liu and Muse 2005). The admixture model of STRUCTURE 2.2 software (Pritchard et al. 2000; Falush et al. 2003) was used to detect the population structure and to assign individuals to subpopulations. The optimum number of populations (K) was selected after five independent runs of a burn-in of 100,000 iterations followed by 250,000 Markov Chain Monte Carlo iterations testing from K = 1 to K = 10. The optimal value of K was estimated by calculating ΔK according to the method of Evanno et al. (2005), where ΔK = m(|L(k + 1) − 2 L(k) + L(k − 1)|)/s[L(k)], and L(k) represents the kth LnP(D), m is the mean of 10 runs and s is their standard deviation.
Linkage disequilibrium was evaluated for each pair of SSR loci using the TASSEL standalone program (ver. 2.1) (http://www.maizegenetics.net). The association analyses were also carried out in TASSEL 2.1 according to Yu et al. (2006) and Bradbury et al. (2007). Four models were used to evaluate the effects of population structure. The general linear model (GLM) was employed to identify association between loci and trait not considering population structure; a Q matrix was employed to identify associations between loci and trait considering population structure; and two mixed linear models (K + MLM and Q + K MLM) (Yu et al. 2006) were employed to identify associations between loci and trait considering the kinship (K) matrix and the population structure (Q) matrix. A data file of the K matrix was created in TASSEL 2.1 by calculating pairwise kinship coefficients using the procedures of Loiselle et al. (1995) and Ritland (1996). The significance of associations between loci and the trait of interest was described as an adjusted P value, and the effects of genotypic variation were used for GLM or for QGLM by individual regression while multiple regressions was used for KMLM and for Q + KMLM estimated by regression (R) 2.
Results
Phenotypic analysis of DMR
The descriptive statistics of the DM scores are summarized in Table 3. The DM scores for the NCSRC-IICRD KU location ranged from 0 to 100 with an average of 32.9, while the DM scores for the NFR location ranged from 0.3 to 100 with an average of 44.5. Histograms of DM ratings of the 60 genotypes did not follow a normal distribution at either of the two locations (Fig. 1a, b). Mean squares values were calculated from an analysis of variance of DM scores of the 60 inbred lines at each location.

Comparison of distribution of % downy mildew for number maize inbred lines at NCSRC-IICRD KU (a) and NFR (b) locations. Percentage disease was calculated as the ratio of the number of systemically infected plants to the total number of plants multiplied by 100. DM ratings are described in "Materials and methods" section
The ANOVA showed significant variation among entries (Table 4). There was a significant location effect as well as a significant entry × location effect. The results from the analysis of variance were used to measure broad sense heritability. Broad sense heritability estimated at each individual location was 0.95 at NCSRC-IICRD KU and 0.97 at NFR. These values indicated a low level of error in evaluating the phenotypic value by scoring three times in each location. Broad sense heritability across locations was 0.97 (highly heritable), suggesting that selecting for this trait would be not difficult.
Population structure
The panel consisted of how many SSR markers were used to identify grouping of maize inbred lines. The 48 markers produced a total of 489 alleles among 60 entries. Alleles per locus varied from 2 to 29, with an average of 10.1. The average PIC (polymorphic information content) of these markers was 0.67 (with a range of 0.19–0.94). The average genetic diversity was 0.70 with a range of 0.19–0.93 (Table 2).
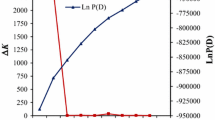
Population structure and genetic diversity of germplasm are shown in Fig. 2. The results showed a steadily increasing curve for LnP(D), indicating that the posterior probabilities for the number of subpopulations increased steadily. The method of Evanno et al. (2005) indicated the value of ΔK was highest at K = 2 (Fig. 2). A dendrogram was generated using the UPGMA algorithm and the GD matrix to create a neighbor-joining tree. All of the entries could be grouped into two subpopulations (Fig. 3). In this dendrogram, subpopulation one mainly consisted of field corn and two lines of popcorn. Subpopulation two comprised all the sweet corn, four lines of waxy corn and some lines of field corn (Fig. 3). Pairwise kinship estimates based on SSR data showed that 49.5 % of the kinship values ranged from 0 to 0.35 (Fig. S1). These results indicated that inbred lines in this study have a moderate level of kinship.

Estimation of the number of subpopulations plotted by ΔK based on posterior probabilities

Dendrogram representing the relationship between the maize inbred lines as revealed by neighbor-joining tree analysis, based on Nei (1983) using the PowerMarker 3.25 program
Because DM ratings varied between locations, association analyses were evaluated for each location separately. Marker-trait associations were identified for 3 SSRs: bnlg1057 on chromosome 1 (bin1.06); bnlg1138, on chromosome 2 (bin 2.06); and umc1033, on chromosome 9 (bin 9.02). These genomic regions conferring DM resistance were detected at the 5 % probability level with the GLM, Q + GLM, K + MLM and Q + K MLM models. R 2 ranged from 28.9 to 72.3 % at NCSRC-IICRD KU (Table 5). Three SSRs were also significant in the K + MLM and Q + K MLM models; however, no significant association was found in the GLM and Q + GLM models for the bnlg1138 locus at the NFR location.
Discussion
Susceptibility to DM infection in maize is a complex trait controlled by both genetics and environment. Symptoms vary depending on plant age and climatic conditions (Frederiksen and Renfro 1977). In this study, mean square of location was significantly higher than entry and entry (G) × location (L) effects for downy mildew. However, the total variability was partitioned among the five factors of variation. The expected mean square of entry (1136.39) was the highest when comparing with location (194.08) and entry × location (58.41), respectively. The results indicate a small G × E for downy mildew. High entry effect indicates for downy mildew that entries are more diverse and sufficient for association studies of downy mildew because of large genotypic effects.
The effectiveness of association mapping depends on the choice of germplasm, correct estimation of population structure, precise trait evaluation, successful identification of candidate polymorphisms (or those linked to the causal polymorphism), and proper statistical analysis (Flint-Garcia et al. 2005). Although our sample was restricted to 60 inbred lines, the average genetic diversity and number of alleles (0.7 and 10.1, respectively) was comparable to previous reports (Yang et al. 2010; Wang et al. 2008), but lower than the highest of 21.7 alleles per locus over 94 SSR loci reported in 260 inbred lines from the US (Liu et al. 2003).
In association mapping, understanding population structure is important for reducing confounding effects and avoiding spurious associations between phenotype and genotype (Pritchard and Rosenberg 1999; Pritchard et al. 2000). Using STRUCTURE to estimate the population structure (Q matrix) (Pritchard et al. 2000; Falush et al. 2003), expressed as membership probabilities, is one way to control for the false positives (Type I errors) caused by population structure creating spurious associations due to familial genetic relatedness (Flint-Garcia et al. 2005) and this has been done in numerous studies (Wang et al. 2008; Xie et al. 2008; Yang et al. 2010). Tropical and temperate subgroups are easily revealed via marker studies, and complex familial relationships can also be identified. In this study, we were able to group maize inbred lines into 2 groups typified by field corn and sweet corn. However, false positives can arise in situations where the statistical test is valid and the association exists, but it is an association with population structure rather than an association with the trait of interest (Ersoz et al. 2009). Therefore, Yu et al. (2006) suggested incorporating pairwise kinship (the K matrix) into a mixed model to correct for relatedness in association mapping. The K matrix is generally superior to association models using only the Q matrix (Yu et al. 2006; Myles et al. 2009). We found that after accounting for the K model and Q + K model using MLM, 3 marker-trait associations for DM resistance still remained significant (α < 0.05) (Table 5) at NCSRC-IICRD KU. At NFR, the non-significant marker-trait associations (P > 0.05) were bnlg1057 and bnlg1138 using the GLM model and the bnlg1138 for the Q GLM model. However, these associations became significant when using the K MLM and Q + K MLM models. Therefore, these marker-trait associations are not false associations caused by population substructures or relatedness among individual lines. These results indicate a moderate kinship effect on association analysis in this population. In the present study, using only 48 SSR markers, we observed only weak intra- and interchromosomal LD.
The genetics of DM resistance in maize can be complex and dependent on genetic background. In this study, we have identified three new SSR markers associated with DM resistance that do not correspond to the previous studies (George et al. 2003; Nair et al. 2005; Sabry et al. 2006; Jampatong et al. 2008). However, all studies found chromosomes 2 carried resistance allele. But resistance allele was located on different bin locations. It is possible that there are diverse alleles affecting DM resistance that were not detected in bi-parental crosses but could be detected using AM procedures. The detection of significant markers both in AM and linkage analysis validates the QTL for DM resistance and suggests that corresponding QTL are present in some other lines of the collection in this study. Future linkage analysis can help to confirm these findings, and bi-parental QTL mapping populations should be created using lines containing the resistant alleles for the three SSR markers identified here. The results from this research will be helpful for breeders to choose parents for crossing and markers for the use of MAS in maize breeding, especially following validation of these resistance alleles in independent studies.
References
Agrama HA, Moussa ME, Naser ME, Tarek MA, Ibrahim AH (1999) Mapping of QTL for downy mildew resistance in maize. Theor Appl Genet 99:519–523
Benbouza H, Jacquemin JM, Baudoin JP, Mergeai G (2006) Optimization of a reliable, fast, cheap and sensitive silver staining method to detect SSR markers in polyacrylamide gels. Biotechnol Agron Soc Environ 10:77–81
Borges OL (1987) Diallel analysis of maize resistance to sorghum downy mildew. Crop Sci 27:178–181
Bradbury PJ, Zhang ZW, Kroon DE, Casstevens RM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23(19):2633–2635
Buckler ES, Gore M (2007) An Arabidopsis haplotype map takes root. Nat Genet 39:1056–1057
Buckler ES, Thornsberry JM (2002) Plant molecular diversity and applications to genomics. Curr Opin Plant Biol 5:107–111
Dalmacio S (2000) Importance of and growing concern for maize diseases in the Asian region. In: Vasal SK, Gonzalez Ceniceros F, Fan XM (eds) Proc 7th Asian regional maize workshop. PCARRD, Los Baños, pp 267–276
De Leon C, Ahuja VP, Capio ER, Mukherjee BK (1993) Genetics of resistance to Philippine downy mildew in three maize populations. Indian J Genet 4:406–410
Don RH, Cox PT, Wainwright BJ, Baker K, Mattick JS (1991) ‘Touchdown’ PCR to circumvent spurious priming during gene amplification. Nucleic Acids Res 19:4008
Doyle JJ, Doyle JL (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Ekasingh B, Gypmantasiri P, Thong-ngam K, Grudloyma P (2004) Maize in Thailand: production systems, constraints, and research priorities. CIMMYT, Mexico
Ersoz ES, Yu J, Buckler ES (2009) Applications of linkage disequilibrium and association mapping in maize. In: Kriz AL, Larkins BA (eds) Molecular genetics approaches to maize improvement, biotechnology in agriculture and forestry, vol 63. Springer, Berlin
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Exconde OR, Raymundo AD (1974) Yield loss caused by Philippine corn downy mildew. Philipp Agric 58:115–120
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587
Fehr WR (1987) Principles of cultivar development, volume 1: theory and technique. McGraw Hill Inc, New York
Flint-Garcia SA, Thornsberry JM, Buckler ES (2003) Structure of linkage disequilibrium in plants. Annu Rev Plant Biol 54:357–374
Flint-Garcia SA, Thuillet AC, Yu JM, Pressoir G, Romero SM, Mitchell SE, Doebley J, Kresovich S, Goodman MM, Buckler ES (2005) Maize association population: a high-resolution platform for quantitative trait locus dissection. Plant J 44:1054–1064
Frederiksen RA, Renfro BL (1977) Global status of maize downy mildew. Annu Rev Phytopathol 15:249–275
George ML, Prasanna BM, Rathore RS, Setty TA, Kasim F, Azrai M, Vasal S, Balla O, Hautea D, Canama A, Regalado E, Vargas M, Khairallah M, Jeffers D, Hoisington D (2003) Identification of QTLs conferring resistance to downy mildews of maize in Asia. Theor Appl Genet 107:544–551
Jampatong C, Jampatong S, Balla C, Grudloyma P, Jompuk C, Prodmatee N (2008) QTL mapping for downy mildew (Peronosclerospora sorghi) resistance in maize. In: Maize for Asia: emerging trends and technologies of the 10th Asian regional maize workshop. Makassar, pp 291–298
Jeffers D, Cordova H, Vasal S, Srinivasan G, Beck D, Barandiaran M (2000) Status in breeding for resistance to maize diseases at CIMMYT. In: Vasal SK, Gonzalez Ceniceros F, Fan XM (eds) Proc 7th Asian regional maize workshop. PCARRD, Los Baños, pp 257–266
Kaneko K, Aday BA (1980) Inheritance of resistance to Philippine downy mildew of maize, Perenosclerospora philippinensis. Crop Sci 20:590–594
Liu KJ, Muse SV (2005) PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21:2128–2129
Liu K, Goodman MM, Muse SV, Smith JS, Buckler ES, Doebley JF (2003) Genetic structure and diversity among maize inbred lines as inferred from DNA microsatellites. Genetics 165:2117–2128
Loiselle BA, Sork VL, Nason J, Graham C (1995) Spatial genetic structure of a tropical understory shrub, Psychotria oYcinalis (Rubiaceae). Am J Bot 82:1420–1425
Myles S, Peiffer J, Brown PJ, Ersoz ES, Zhang ZW, Costich DE, Buckler ED (2009) Association mapping: critical considerations shift from genotyping to experimental design. Plant Cell 21:2194–2202
Nair SK, Prasanna BM, Garg A, Rathore RS, Setty TAS, Sing NN (2005) Identification and validation of QTLs conferring resistance to sorghum downy mildew (Peronosclerospora sorghi) and Rajasthan downy mildew (P. heteropogoni) in maize. Theor Appl Genet 110:1384–1392
Nei M (1972) Genetic distance between populations. Am Nat 106:283–292
Nei M (1983) Genetic polymorphism and the role of mutation in evolution. In: Nei M, Koehn R (eds) Evolution of genes and proteins. Sinauer Associates, Sunderland, pp 165–190
Phumichai C, Doungchan W, Puddhanon P, Jampatong S, Grudloyma P, Kirdsri C, Chunwongse J, Pulam T (2008) SSR-based and grain yield-based diversity of hybrid maize in Thailand. Field Crops Res 108:157–162
Pritchard JK, Rosenberg NA (1999) Use of unlinked genetic markers to detect population stratification in association studies. Am J Hum Genet 65:220–228
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Raymundo AD (2000) Downy mildew of maize in Asia: new perspectives in resistance breeding. In: Vasal SK, Gonzalez Ceniceros F, Fan XM (eds) Proc 7th Asian regional maize workshop. PCARRD, Los Baños, pp 277–284
Ritland K (1996) Estimators for pairwise relatedness and individual inbreeding coefficients. Genet Res 67:175–186
Sabry A, Jeffers D, Vasal SK, Frederiksen R, Magill C (2006) A region of maize chromosome 2 affects response to downy mildew pathogens. Theor Appl Genet 113:321–330
Singburaudom N, Renfro BL (1982) Heritability of resistance in maize to sorghum downy mildew Perenosclerospora sorghi (Weston and Uppal) C. G. Shaw. Crop Prot 1:323–332
Sriwatanapongse S, Jinahyon S, Vasal SK (1993) Suwan-1: maize from Thailand to the world. CIMMYT, Mexico
Stich B, Melchinger AE, Frich M, Maurer HP, Heckenberger M, Reif JC (2005) Linkage disequilibrium in European elite maize germplasm investigated with SSRs. Theor Appl Genet 111:723–730
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Thornsberry JM, Goodman MM, Doebley J, Kresovich S, Nielsen BD, Buckler ES (2001) Dwarf8 polymorphisms associate with variation in flowering time. Nat Genet 28:286–289
Wang RH, Yu YT, Zhao JR, Shi YS, Song YC, Wang TY, Li Y (2008) Population structure and linkage disequilibrium of a mini core set of maize inbred lines in China. Theor Appl Genet 117:1141–1153
Weir BS (1996) Genetic data analysis II. Sinauer Associates Inc., Sunderland
Xie CX, Warburton M, Li MS, Li XH, Xiao MJ, Hao ZF, Zhao Q, Zhang SH (2008) An analysis of population structure and linkage disequilibrium using multilocus data in 187 maize inbred lines. Mol Breed 21:407–418
Yang XH, Guo YQ, Yan JB, Zhang J, Song TM, Rocheford T, Li JS (2010) Major and minor QTL and epistasis contribute to fatty acid composition and oil content in high-oil maize. Theor Appl Genet 120:665–678
Yu J, Buckler ES (2006) Genetic association mapping and genome organization of maize. Curr Opin Biotechnol 17:155–160
Yu J, Pressoir G, Briggs WH, Vroh Bi I, Yamasaki M, Doebley JF, McMullen MD, Gaut BS, Nielsen DM, Holland JB, Kresovich S, Buckler ES (2006) A uniWed mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet 38:203–208
Zhu CS, Gore M, Buckler ES, Yu JM (2008) Status and prospects of association mapping in plants. Plant Genome 1:5–20
Acknowledgments
The authors gratefully acknowledge Marilyn Warburton for useful comments for improvement of the manuscript. This research was funded by the Generation Challenge Programme (GCP) and the National Centre for Genetic Engineering and Biotechnology (BIOTEC).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Phumichai, C., Chunwongse, J., Jampatong, S. et al. Detection and integration of gene mapping of downy mildew resistance in maize inbred lines though linkage and association. Euphytica 187, 369–379 (2012). https://doi.org/10.1007/s10681-012-0699-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10681-012-0699-8