
Abstract
Barley (Hordeum vulgare) is a major feed source for the intensive livestock industry. Competitiveness against other cereal grains depends largely on the price per unit of expressed feed quality. The traits which contribute to feed quality in barley are largely quantitative in nature but little is known about their genetic control. A study to identify the quantitative trait loci (QTLs) associated with feed quality was performed using a F6-derived recombinant inbred barley population. Samples from each line were incubated in the rumen of fistulated cattle, recovered, washed and dried for determination of in situ dry matter digestibility. Additionally, both pre- and post-digestion samples were analysed to quantify the content of key quality components relating to acid detergent fibre, total starch and protein. The data was used to identify trait-associated QTLs. Genetic analysis identified significant QTLs on chromosomes 2H, 5H and 7H. Genetic markers linked to these QTL should provide an effective tool for the selection and improvement of feed barley in the future.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In order to measure the nutritional value of absorbed feedstuff in animals, digestibility of the feed source must be determined. In ruminants such as cattle, this is frequently expressed as dry matter digestibility (DMD), which can be determined by several methods. The first, in vivo, is characterised by feeding animals with known amounts of sample and measuring faecal output (Matsushima 1979). Although in vivo analysis is accurate and has been extensively used, it is expensive and requires a large amount of grain for the evaluation of a small number of samples (Khorasani et al. 2000). The second method, an in vitro approach, determines digestibility by simulating ruminal chemistry and enzymatic reactions (McDonald et al. 1995). While being rapid and relatively inexpensive, it does not replicate the actual ruminal physiology, resulting in lower digestibility estimates when compared to in vivo measurements. The third method is an in situ technique, which involves the insertion of multiple non-digestible porous nylon bags, filled with grain samples, into the rumen of cannulated animals. This allows the amount of dry matter digested within the bag to be measured over a set period (Herrera-Saldana et al. 1990; Khorasani et al. 2000) and provides data from within the rumen itself. Ruminal digestion of incubated samples in the nylon bag can be affected by a number of factors, such as bag and sample size, bag material, pore size, sample processing, animal diet, feeding level and frequency, bag insertion and removal procedures, location of the bag in the rumen, rinsing procedure, microbial correction and incubation time. Bowman et al. (2001) formulated a standardised procedure for DMD evaluation and observed genotypic effects on in situ DMD, indicating that improved feed quality could be achieved by selecting specific quality traits.
DMD results indicate which samples have the greatest degree of digestibility, but do not identify nor quantify which nutrients were digested. Both the physical and chemical properties of cereal grains and their interactions influence performance and ruminal digestion which notably affects the degree of digestion and available energy. With no consensus as to what constitutes feed quality, animal performance was traditionally used and measured by average daily weight gain. Surber et al. (2000) identified four basic traits that constitute and affect barley feed quality traits; acid detergent fibre (ADF), starch, protein and DMD. In order to identify quantitative trait loci (QTLs) associated with these quality traits, DMD and nutrient content need to be determined by analytical chemistry.
In this study we determine the nutritional content in whole barley grain and the digested residue and DMD of a number of lines of a Tallon × Scarlett recombinant inbred population. Phenotypic data from the in situ trial was used to identify pre- and post-digestion QTLs. These QTLs of interest were specifically associated with feed quality traits within the Tallon × Scarlett population.
Materials and methods
Barley samples
An F6-derived recombinant inbred (RI) population, derived from a cross between Tallon and Scarlett, comprising 140 individuals (Lawson et al. 2009), was used to estimate in situ digestibility and for the identification of QTL associated with feed quality traits. Both Tallon and Scarlett are malting varieties that also exhibit desirable feed traits (Fox et al. 2009). Tallon, a cross between Triumph and Grimmett, is a malting variety developed by the former Queensland Department of Primary Industries, Hermitage Research Station, Warwick, Queensland, Australia, while Scarlett is a European malting variety produced by a Amazone/Breun ST 2 730E//Kym cross.
Dry matter digestibility
Seven 3-year-old rumen-fistulised Bos taurus cross-bred steers with average weight of 479.3 kg (standard error of 11.8 kg) were selected for the in situ trial; and housed in individual pens at the Centre for Advanced Animal Studies (CAAS), The University of Queensland, Gatton, Queensland, Australia. The steers were weighed and individual rations calculated to transfer animals to the required 1:1 grain/hay diet in preparation for the trial. Average daily weight gain was recorded to ensure animal welfare, performance and compliance with animal ethics requirements.
The study used a partially duplicated design using grain from 200 field plots comprised of 140 Tallon and Scarlett F6-derived RI lines as treatments, as implemented in the design package DiGGer (Coombes 2002). This formed the second stage of testing on this material following grain production during field trials. As such, it was crucial for experimental purposes to be able to estimate differences between grain samples from the field, and those from animal testing, in order to identify the genetic variance. Grain from field plots were allocated randomly to six animals per day (incomplete blocks), where each animal contained 30 bagged samples of grain. This formed a total of 180 bags per day, ensuring that all 140 genotypes were tested daily with an additional 40 samples forming partial duplication of the field plots. Five days of sampling occurred forming five complete replicates of the genotypes. A total of 900 bags were assessed, while a seventh animal was prepared as a potential replacement, should a participating animal be taken ill and have to be removed from the trial.
Grain samples were prepared by rolling them through a brewer’s hand mill (Schnitzer’s CAMPO Flockenquetsche) with a 2 mm gap size. Approximately 5 g was placed in numbered French stitched Monofilament polyester bags (24 × 10 cm2) with pore size of 45 μm. Prior to use, the empty bags were placed in a drying oven at 55°C for 24 h and the empty bag weight was recorded. Bag number, sample and combined (bag and sample) weights were recorded. Grain-filled sample bags were sealed and stored in dry airtight containers marked according to the day of incubation and corresponding animal. During sample preparation sub-samples were taken. The subsamples were weighed and then dried at 105°C for 48 h, before being weighed again and moisture content calculated.
The fistular bags were attached to a chain and soaked for 3–5 min in clean water prior to incubation. The animals were secured in a crush to allow safe and easy access to the cannula where the chain and bags were inserted, and pushed down below the feed raft. This ensured maximum exposure to ruminal microbes and digestive enzymes. Samples were digested in situ for 3 h before removal. After removal, external ruminal residue was removed before the samples were immersed in cold water to prevent further enzymatic digestion. After washing under cold water, the bags were dried in a force-draught oven at 55°C for 48 h. The dried samples were weighed before being stored in labelled screw top jars for post-digestion nutritional analysis. The DMD of each line was expressed as a percentage of the dry sample residue weight relative to the initial dry sample weight.
Nutritional analysis
Whole grain barley samples (‘pre-digestion’) and the post-digestion residues were ground prior to protein, ADF and starch analysis. Samples were ground using a Foss-Tecator, Cyclotec 1093 mill to pass through a 1 mm screen. The pre- and post-digestion barley flour was analysed for protein content using a Vario Max N/CN Macro elemental analyser as per manufacturers’ specification. Two runs for each sample were performed, with data recorded for statistical analysis and comparison. The ADF content was determined for both the pre- and post-digestion samples using the Fibre Tec 2021 Fibrecap system (Soest 1994). The Megazyme total starch analysis kit was used on pre- and post-digestion samples as per manufacturer’s specifications to determine the starch content.
A linear mixed model was fitted to the nutritional data with terms for genotype as the primary effect of interest, and blocking effects of days and animals fitted as random terms. Residual maximum likelihood (REML) was used to give estimate variance components (Patterson and Thompson 1971). All nutritional data was analysed using the software package ASReml-R (Butler et al. 2009). Associated bi-plots of genotype by trait means were constructed to explore the relationships between the nutritional traits.
QTL identification
A genetic linkage map for the F6-derived Tallon × Scarlett RI population was provided by the DEEDI Barley Breeding program based at Hermitage Research Facility, Warwick, Queensland. The provided molecular map has a total distance of 1224.48 cM, with an average distance of 5.3 cM between the markers. The map consisted of 14 linkage groups for the seven chromosomes, including three linkage groups for chromosomes 1H and 7H, and two linkage groups for chromosomes 2H, 5H and 6H. Gaps in the linkage groups are most likely due to regions of markers that were not polymorphic between the two parents.
Composite interval mapping (CIM) was used to detect QTLs and estimate the magnitude of their effects, using the stepwise regression analysis in WinQTLCartographer 2.0 (Wang et al. 2002). The percentage of phenotypic variance (R 2) explained by a specific QTL was taken at the peak of the QTL. A series of 1,000 permutations were run to determine the experiment-wise significant level at P = 0.05 of logarithm of odds (LOD) for the trait (Churchill and Doerge 1994). Permutation tests indicated that a LOD score of 2.7 or greater was significant and showed linkage to a quality trait. QTLs were graphically represented using MapChart 2.2 (Voorrips 2002). QTLs have been named according to the nomenclature style described on GrainGenes (http://wheat.pw.usda.gov/GG2) whereby the name consists of a Q: denoting a QTL, a 2–4 letter acronym describing the trait, an abbreviation of the genetic cross and the chromosome.
Results
DMD and nutritional data
The population distribution for percentage dry matter digested following the 3 h in situ incubation was varied, ranging from 43% through to 68% digestion in the individual lines. Transgressive segregation for both higher and lower digestibility, compared with the parents, was observed (Fig. 1). ADF digestion/loss ranged from approximately 2% through to 8%, while the percentage protein and starch digested, as derived by chemical analysis and calculations, yielding distributions ranging from 24% through to 68% and 36% through to approximately 82% respectively (Fig. 1). Transgressive segregation was also clearly observed for both higher and lower %ADF content, %starch digested and %protein digested when compared to both parents (Fig. 1).

The phenotypic distribution of the percentage DMD, starch, protein and ADF digested after 3 h in situ incubation
The statistical analysis of the data estimated variance components for each random term in the model. Genotype (Comp = 10.2%) had a minor contribution to the total variance observed, when experimental and environmental factors were taken into consideration. The analysis indicated experimental error (Comp = 33.7%) had the greatest contribution to the observed variance, followed by the day the trial was performed (Comp = 21.0%), and by day/animal interactions (Comp = 20.1%) (Table 1).
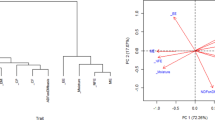
The highest contribution to the observed variation in protein content across the sample population arose from field-plot effects rather than genotype. The relatively low contribution of genotype to protein content variance may be attributed to the shared pedigree of the parental lines. The relationship between ten feed quality traits (DMD; pre-digestion starch, protein, ADF; post-digestion starch, protein, ADF and starch, protein, ADF digested) was analysed using principal component analysis (Fig. 2). Principal component 1 (PC1) and component 2 (PC2) explained 46% of the total observed variance, with PC1 accounting for 25% of the observed variance and PC2 21%, respectively. Significant (P < 0.05) genetic correlations were identified between pre- and post-digestion protein content (r = 0.639), pre-digestion starch content and quantity of starch digested (r = 0.655) and between post-digestion ADF (remaining residue) and ADF digested (r = 0.954). A negative correlation was found between post-digestion starch content and the amount of starch digested (r = −0.722). A negative correlation was also observed between pre-digestion protein concentration and DMD (r = −0.537). Protein digested and pre-digestion starch content are also correlated to each other, while both are negatively correlated to post-digestion ADF content and ADF digested. Numerous other moderate and weak correlations were also identified (Table 2; Fig. 2).

Bi-plot representing 46% variance of the data set explained. The variance of the bi-plot is broken down into two components found on the axis representing 25% (X-axis) and 21% (Y-axis). The correlation between two points observed is derived by the determination of the cosine of θ. The distance between the two points is interpreted as the variance between the points
With ADF, the main source of variance is experimental error (error variance), which is relatively small and of the same magnitude for both pre- (Var. = 0.023) and post-digestion (Var. = 0.012). In protein, the main source of variance observed was contributed by genetic variance with the pre-digestion (Var. = 0.98) and post-digestion variance (Var. = 1.062) being of similar magnitude.
Identification of feed quality QTLs
Genetic analysis of total digested protein identified a QTL on chromosome 2H between the DArT markers bPb-1611 and bPb-6296, and explained 13% of the phenotypic variance. In addition, genetic analysis identified two QTL for DMD on chromosomes 5H and 7H. The QTL on chromosome 5H, located between the flanking DArT markers bPb-8072 and bPb-1820, explained 19% of the phenotypic variance, while the QTL on 7H was located between flanking markers bPb-1767 and bPb-0758 and explained 9.9% of the phenotypic variance. The QTLs on 2H and 5H were contributed by Tallon, whereas the QTL on 7H was contributed by Scarlett. A number of suggestive QTL for pre-, post-digestion protein and digested protein were also identified (data not shown) with LOD scores ranging from 2.1 to 2.4. These QTLs explained between 5.7 and 14.4% of the phenotypic variance. Suggestive QTL (LOD < 2.7) for DMD were identified on chromosomes 1H, 3H, 5H, 6H and 7H and explained between 5.9 and 9.1% of the phenotypic variance (Table 3; Fig. 3).

QTL analysis for feed quality traits in Tallon × Scarlett F6-derived RIL population
Discussion
Research in Australia on barley has traditionally focused on agronomic traits, disease resistance and malting with little input into the genetic factors affecting feed quality (Anderson and Reinbergs 1985). With recent research identifying chemical and physical factors such as DMD, starch, protein and fibre (ADF) as important feed quality traits, greater emphasis is now placed on breeding for these characteristics. In this current study a mapped F6-derived RI line population of Tallon × Scarlett has been employed to determine ADF content, protein digestion, starch digestion and DMD through in situ trials conducted in rumen-fistulised B. taurus cattle. It was the first time that a study assessing grain feed quality attributes for QTL analysis was undertaken in an Australian cattle herd. Previously, herds in the US have been used in phenotypic analysis of Australian grain samples. However, different herds and breeds can introduce additional genotypic variances. Additionally if separate QTLs were identified for pre-and post-digestion traits is this an indication of genetics of the grain influencing the digestibility of the trait? Or is the genetics of the herd used and their ability to digest the aforementioned trait responsible for this variation? However with appropriate statistical designs and analyses, the genotypic differences between herds can be minimised.
Phenotypic results from in situ digestion on the Tallon × Scarlett population showed transgressive segregation for feed quality traits. These distributions suggest that feed quality trait expression was controlled by multiple genes derived from both parents. The phenotypic data showed a distinct correlation between the measured feed traits. Correlation analysis was performed on the data set where known sources of variance were statistically manipulated. Principal component analysis explained only 41% of the variance. This may have partially been due to the correlations being weighted rather than an error with the experimental design or data. The weighting of the correlations may be explained by the limited removal of variance from the data set due to the lack of experimental replication in fibre (ADF) and starch analysis caused by financial constraints. This resulted in the attributable/explained variance being manipulated for only half the feed traits with sufficient replications. This allowed the genetic variance to be determined for trait data with sufficient replications. Nevertheless the correlations observed have increased understanding of the feed quality traits and their interactions during ruminal digestion.
Analysis of the genetic map of the Tallon × Scarlett population allowed the detection of three significant QTLs linked to the two feed quality traits, protein digested and DMD. These QTLs mapped to chromosomes 2H, 5H and 7H. Tallon had the greatest contribution to feed quality traits in the population tested. The significant QTL linked to DMD on 5H in this study also corresponds to a QTL for DMD reported by Lawson et al. 2009. Identification of QTLs within the same genomic region as previously reported validates the QTLs that are consistently detected across populations and environments as important regions associated with feed quality. Similar studies found QTL associated with feed quality on the same chromosomes as the present study’s suggestive QTL on chromosomes 1H, 3H, 5H, 6H and 7H. These suggestive QTL are primarily linked to DMD and protein. An exciting prospect of the current research was the ability to detect separate suggestive QTL associated feed quality traits pre- and post-digestion as well as digested. This can best be illustrated by a suggestive QTL linked to pre- and post-digestion protein on chromosome 1H with flanking markers bPb-3660 and CTC206 and the suggestive QTL for protein digested on chromosome 1H with flanking markers fGA/CCC 137–bPb-3984. Despite the fact that we were able to differentiate between pre- and post-digestion QTL, additional investigations need to be performed to validate these findings. The aim of the experiment was not just to identify QTLs associated with feed quality, but also to help establish efficient and reliable protocols for future research of this nature in Australia. Identification of common QTLs also helps to validate the experimental resources locally available as these factors will contribute to variation of QTL location and significance.
The feedlot, dairy and chicken industries have driven feed grain demand in Australia from 5.7 million tonnes in 1993 to 11.9 million tonnes in 2007, with an average increase of 4.1% each year (Martin et al. 2009; Roberts et al. 2009). At present, barley is the second most widely produced cereal crop in Australia, with an average production of approximately 4.1 million tonnes of feed barley, and a domestic consumption of about 2 million tonnes per annum (Rebetzke 2007; Martin et al. 2009). The domestic demand for barley is driven mainly by the livestock industry (Smith 1995) which uses cereal grain as a primary energy source. Despite being the preferred grain due to its energy availability, relative price and availability of barley determines its competitiveness against other grains in this market.
A greater consensus in the feed industry as to what defines feed quality is essential. Once a clear definition of feed quality is agreed upon amongst plant breeders and other industry players, the individual traits and the intricate interactions between these traits can be studied in detail. Such data would encourage more detailed mapping studies. From the present research, the putative QTLs derived from Tallon and Scarlett indicate it may be necessary to run molecular markers across their individual pedigrees to potentially identify additional QTLs linked to feed quality traits. An increased number of QTLs identified and mapped may lead to the breeding and growing of an industry-specific barley crop with a higher energy and nutrient content. Additional markers need to be incorporated in genomic regions where feed quality QTLs have been reported in order to increase map density and tighten linkage between the markers and unidentified genes controlling the trait in question. This would facilitate and promote marker assisted selection in breeding programs targeted to feed quality. Phenotyping methodology refinement, the discovery and development of new genetic techniques and the use of a larger sample population may help validate some of the suggestive markers found in this study.
Conclusions
It is possible for the genetic components in studies centred on the use of fistulated cattle to be dissected by appropriate experimental designs. These designs could potentially eliminate the impact of attributable sources of variance such as animal, day, experimental error and day/animal effects; thus highlighting the observed genotypic effects. These statistical approaches can be confidently applied to barley feed quality QTL analysis. Potentially useful markers exist, which define these critical QTL and could be used in the genetic improvement of barley feed quality. The molecular markers defining these QTL will be effective tools for selection and genetic improvement of feed barley in the future. The significant contributions that environmental conditions, animals and their subsequent interaction with each other make to variance, render it essential to perform these studies under local conditions. The use of established protocols that were refined and used in this study has made it is possible to perform complex feed quality trait analysis, under Australian conditions in adapted local cattle breeds.
References
Anderson MK, Reinbergs E (1985) Barley breeding. Barley agronomy monograph, 26th edn. American Society of Agronomy, Madison
Bowman JGP, Blake TK, Surber LMM, Habernicht DK, Bockelman H (2001) Feed quality variation in the barley core collection of the USDA national small grains collection. Crop Sci (41):863–870
Butler DG, Cullis BR, Gilmour AR, Gogel BJ (2009) ASReml-R reference manual release, 3 edn. Queensland Department of Primary Industries, Technical report
Churchill GA, Doerge RW (1994) Empirical threshold values for quantitative trait mapping. Genetics 138:963–971
Coombes NE (2002) The reactive tabu search for efficient correlated experimental designs. Liverpool John Moores University, Liverpool
Fox G, Kelly A, Bowman J, Inkerman A, Poulsen D, Henry R (2009) Is malting barley better feed for cattle than feed barley? J Inst Brew 115(2):95–104
Herrera-Saldana RE, Huber JT, Poore MH (1990) Dry matter, crude protein and starch degradability of five cereal grains. J Dairy Sci 73:2386–2393
Khorasani GR, Helm J, Kennelly JJ (2000) In situ rumen degradation characteristics of sixty cultivars barley grain. Can J Anim Sci 80:691–701
Lawson W, Mace E, Collard BCY, Fox G, Kelly A, Sutherland M, Franckowiak J, Bowman J (2009) Investigating the molecular dynamics of feed quality traits in two barley F6-derived RIL populations. Paper presented at the Australian Plant Breeding Conference, Cairns
Martin P, Mackinnon D, Dharma S, Liao B, Phillips P, Crooks S, Penm J (2009) Australian farm survey results. Australian Bureau of Agricultural and Resource Economics
Matsushima JK (1979) Feeding beef cattle. Advances series in agriculture sciences. Springer, New York
McDonald P, Edwards RA, Greenhalgh JFD, Morgan CA (1995) Animal Nutrition, 5th edn. Longman Scientific and Technical, New York
Patterson HD, Thompson R (1971) Recovery of inter-block information when block sizes are unequal. Biometrika 58:545–554
Rebetzke G (2007) CSIRO: barley overview. CSIRO. 2009
Roberts I, Haseltine C, Maliyasena A (2009) Factors affecting Australian agricultural exports. Australian Bureau of Agricultural and Resource Economics
Smith W (1995) Crop production: evolution, history and technology. Wiley, New York
Soest PJV (1994) Nutritional ecology of the Ruminant, 2nd edn. Cornell University Press, New York
Surber LMM, Bowman JGP, Blake TK, Hinman DD, Boss DL, Blackhurst TC (2000) Prediction of barley feed quality for beef cattle from laboratory analyses. Proc West Sect Am Soc Anim Sci (51):454–457
Voorrips RE (2002) MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered 93:77–78
Wang S, Basten CJ, Zeng Z-B (2002) Windows QTL Cartographer: WinQtlCart V2.0
Acknowledgments
The authors acknowledge the Grain Research and Development Corporation (GRDC) and the Department of Employment, Economic Development and Innovation (DEEDI) for funding the project. We thank Donna Hocroft (DEEDI) and Jim Kidd (CAAS) for all their time and aid in sample preparation for the phenotypic trial and for maintaining the cattle herd.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gous, P.W., Martin, A., Lawson, W. et al. QTL associated with barley (Hordeum vulgare) feed quality traits measured through in situ digestion. Euphytica 185, 37–45 (2012). https://doi.org/10.1007/s10681-011-0608-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10681-011-0608-6