
Abstract
Sheath blight (SB), caused by Rhizoctonia solani, is a serious disease of cultivated rice (Oryza sativa L.) for which genetic resistance is in demand by breeders. With the goal of resistance (SBR)-QTL discovery in U. S. japonica breeding material, 197 doubled-haploid lines from a cross between MCR10277 (resistant) and Cocodrie (susceptible) were evaluated in field and greenhouse assays with U. S. and Colombian pathogen isolates and genotyped at 111 microsatellite marker loci. Four SBR QTLs from MCR10277 were identified, together accounting for 47% of field genetic variation. In all trials the strongest effect was provided by a chromosome-9 QTL, qsbr_9.1, but some QTLs differed for U. S. and Colombian R. solani isolates. SBR QTLs coincided with only two of several height or heading-time QTLs, suggesting that the relationship between these developmental traits and SBR is not simple. For the U. S. isolates, a microchamber greenhouse assay revealed the same QTLs as did field inoculation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The second most economically important disease of rice in the world (Lee and Rush 1983; Webster and Gunnell 1992) is sheath blight (SB), caused by Rhizoctonia solani Kühn, anastomosis group AG1, intraspecific group IA [teleomorph: Thanatephorus cucumeris (A.B. Frank) Donk.]. Under conditions favoring disease, up to 50% of grain yield may be lost (Marchetti and Bollich 1991). Infection by this necrotrophic pathogen is via soilborne sclerotia that collect around rice culms under flooded paddy conditions, causing initial infection at the waterline and hyphal invasion along the stem to the canopy, where proliferation of water-soaked lesions, corresponding to phytotoxin-killed cells (Brooks 2007) may kill the plant. In the humid southern U. S. rice-growing regions, SB is particularly severe owing to the preferred use of well-tillering susceptible semi-dwarf japonica cultivars planted at high densities, leading to dense canopies with moist microclimates. Rotation with soybean, an alternative host of R. solani, increases the disease pressure (Groth et al. 1991). Control may be achieved with fungicides (Groth 2008), but cost and the potential for development of pathogen resistance make plant genetic resistance preferable.
While no complete SB resistance (SBR) has been found in cultivated or wild rices (Pan et al. 1999), partial resistance is best known in indica rices, where a series of studies in japonica × indica crosses has revealed an assortment of quantitative-trait loci (QTLs) with indica-derived resistance alleles (Channamallikarjuna et al. 2010; Che et al. 2003; Li et al. 1995; Liu et al. 2009; Pinson et al. 2005; Xie et al. 2008; Yin et al. 2009; Zou et al. 2000). However, japonica rices are better adapted to U. S. cultivation (Mackill and McKenzie 2003), prompting the search for useful resistance QTLs within this rice group (Sharma et al. 2009).
For identifying the genetic bases of rice SBR it is desirable to exclude environmental effects by appropriate designs and analytical approaches. Among the indirect influences on phenotypic variation are paddy water depth, nitrogen fertilization rates (Slaton et al. 2003), soil silica content (Rodrigues et al. 2001; Rodrigues et al. 2003), plant growth stage (Liu et al. 2009) and height and heading date (Li et al. 1995; Pan et al. 1999). Tall plant stature favors escape by delaying the pathogen’s arrival in the canopy; short stature at the sd-1 semi-dwarfing locus was strongly linked to higher SB by Sharma et al. (2009). Early or late heading may disrupt the synchrony of plant with pathogen development, giving to otherwise susceptible germplasm the appearance of resistance (Pinson et al. 2005). The correlation of field resistance with controlled-environment disease-screening methods described by Jia et al. (2007) and Liu et al. (2009), which could counter these confounding environmental effects, has not been fully established.
The objectives of this study were to identify sheath-blight resistance QTLs for potential use in japonica backgrounds and characterize their association with plant height and heading time in progeny of a cross between resistant and susceptible U. S. breeding lines evaluated for disease response to two isolates by several inoculation and culture methods.
Materials and methods
Genetic materials
Cocodrie (Linscombe et al. 2000), with pedigree Cypress//L-202/Tebonnet, is a semi-dwarf long-grain cultivar widely grown in the southern U. S. for its high yield potential and good milling yield. It is rated extremely SB-susceptible at 7.7 on the conventional disease-severity scale from 0 (no disease) to 9 (plants dead and collapsed). MCR10277 is an F8 selection from LSBR-5/Lemont//Katy/3/Cypress/Teqing (M. C. Rush, personal communication), with at least two sources of partial resistance in the pedigree: LSBR-5, carrying a single recessive gene identified following tissue culture of susceptible cultivar Labelle (Xie et al. 1992) and Teqing, with a dominant gene (Pan et al. 1999). LSBR-5 is a SB-resistant selection from a field planted to tissue-culture-regenerated lines of susceptible cultivar Labelle and was supposed by Xie et al. (1992) to be a somaclonal variant from Labelle. But in a later genotyping screen of rice accessions, LSBR-5 differed from Labelle alleles at 19 of 24 tested SSR loci, all 19 commonly found in indica-derived germplasm (R. G. Fjellstrom, unpublished data) suggesting that LSBR-5 was an indica-derived outcross or volunteer in the field. MCR10277 also shows good yield potential and is rated at 3 or tolerant. A set of 197 doubled-haploid lines (DHLs) (Chu et al. 2006) (hereafter the SB2 population) developed by anther culture from F1 progeny of these two parents (Cocodrie as the female) was selected for mapping.
Phenotypic evaluation
SB2 was grown in the field at Crowley, Louisiana (LA) in 2005–2007 and Stuttgart, Arkansas (AR) in 2006–2007. Triply replicated 2 m (LA) or 0.6 m (AR)-row plots were sown at 0.25 m row spacing in a randomized complete block design and agronomic practices recommended for each site for plant cultivation and control of weeds and insect pests were applied. Plots were planted in April (LA) or May (AR) and inoculated at the late tillering stage 50–60 days after seeding by spread of inoculum on each row by hand at the rate of 10–15 ml. Rhizoctonia solani isolates used were: in LA, LR172 grown on an moist autoclaved rice-hull: grain (2:1) mixture; in AR, RR0140-1 (Wamishe et al. 2007) grown on a sterilized mixture of corn chops and rye seed (1:1) followed by drying and breaking into small fragments for spreading on plots. Disease severity was evaluated at near maturity for each DHL, in LA about 40 and in AR about 35 days after heading, based on the 0–9 rating scale. Scoring took around three weeks, the range in heading times among the SB2 DHLs.
Plant height was measured at maturity from the soil surface to the top of the extended heads. Heading date was recorded at boot emergence of 50% of heads.
Two other sheath-blight assays were performed under greenhouse conditions: in a mist chamber at the International Center for Tropical Agriculture (CIAT) in Cali, Colombia, and in microchambers (Jia et al. 2007) in Stuttgart, AR. In both assays, the DHLs were grown in 15-cm plastic pots filled with sterile soil and fertilized as described in Liu et al. (2009).
Mist-chamber assay
Rice planting pots, each with five plants of the same DHL or check variety, were placed in a walk-in plastic chamber of dimension 4 × 3 × 2 m with a capacity of 500 pots, at >95% relative humidity maintained by humidifiers and temperature ranging from 21 to 35°C. Pots of 15 cm diam were filled with steam-sterilized soil from the station and fertilized with the equivalent of 180 kg N, 60 kg P, and 60 kg K/ha. Fertilizer application was split in 3 equal amounts: at time of planting, 30 days after planting, and one day before inoculation (50 days after planting). Aggressive Colombian isolate Tol 2399-1 of R. solani AG I-1A was collected, stored, and maintained in the Pathology Laboratory at CIAT as described in Liu et al. (2009). Inoculum was prepared by transfer of actively growing mycelia from potato dextrose agar (PDA) plates to fresh PDA medium followed by culture for 5 days at 27°C in darkness. At 50 days after sowing, mycelial discs (7 mm in diameter) were excised from the culture plate with a pipette tip and placed at the base of each plant in each pot. After incubation in the chamber for 10 days, inoculated plants were transferred to greenhouse benches (21 to 30°C, >80% RH) for 5 days and then returned to the chamber for 5 further incubation days, by which time all flag leaves were well developed. Disease severity was evaluated in percentage of plant area affected (PAA) as follows: a maximum disease severity value of 30% was given to the first (flag) leaf; of 15% to each of the second and third leaves; of 10% to each of the fourth and fifth leaves; and of 20% to the stem for a possible total of 100%. This scale was developed at CIAT based on a high association between %PAA in greenhouse and field performance of a rice cultivar including sheath blight reaction and yield reduction. Each such trial was repeated five times, with pots arranged in a completely randomized design in each trial.
Microchamber assay
Rice was germinated in planting pots and thinned to a maximum of 3 plants per pot. Trays containing eight pots were randomized in the greenhouse for each of two repeated trials, in each of which three replicates of each entry were evaluated. Greenhouse temperatures ranged from 18.3 to 29.6°C and relative humidity from 34 to 81%. R. solani isolate RR0140-1 was cultured and mycelial discs excised as described above. At about 4 weeks after sowing, a disc was placed at the base of each rice seedling and each pot was covered with a 3 l clear plastic bottle whose bottom and cap had been removed, forming a microchamber. At 9–10 days after inoculation the lines were rated as follows: plant height was measured as the length of the leaf sheath from the soil surface to the collar of the most newly emerged leaf, and disease severity score on the standard 0–9 scale was calculated as 9 * (lesion length)/height.
Polymorphism screening and marker genotyping
Of a set of 730 SSR markers screened on the SB2 parents, 318 or 0.44 were found to be polymorphic. Of these, 114 were selected for moderately uniform distribution over the rice genetic map based on positions found in the Gramene database (Ware et al. 2002) and were scored on the DH lines, three being subsequently dropped owing to difficulty in scoring. DNA from parents and each line was isolated from a bulk of five kernels with DNeasy kits (Qiagen) following the manufacturer’s protocol. SSR genotyping followed the protocol of Richards et al. (2004).
Data analysis
Traits
Trait heritabilities were calculated from the field data as described in Dudley and Moll (1969) with an analysis of variance fitting effects of RIL, year, location, and all interactions, as σ 2 g /(σ 2 g + σ 2 gl + σ 2 gy + σ 2 gly + σ 2 ε ) where the g, l, and y subscripts denote genetic (RIL), location, and year effects and the ε error, and variance components were computed from equating observed to expected mean squares (MS). For the micro- and mist-chamber assays heritability was computed analogously as (MS g − MS ε )/[MS g + (r − 1)MS ε ] where the replication number r was respectively 3 and 5.
Linkage and QTL mapping
Marker linkage maps were constructed with CarthaGène (de Givry et al. 2005). For a multiple-environment QTL analysis aimed at separating QTL main from QEI effects, the following model was fitted to the field data with SAS (SAS Institute 2004) PROC MIXED:
where effects (all fixed unless specified as random) are defined as follows: μ the general mean, L j the jth location, Y k the kth year, Q il the lth QTL genotype of the ith RIL, represented by the genotypic expectation at the tested QTL position, calculated with QGene (Joehanes and Nelson 2008) by the method of Jiang and Zeng (1997) at 5-cM intervals on the genetic map and at marker positions; all interaction effects among these variables, B ic the effect of the cth QTL-cofactor genotype for the ith RIL, (fitted as a random effect, though in comparisons this choice proved to make no difference) and ε ijkl the residual variation. This model was fitted at all QTL positions with initial cofactor QTLs selected from multiple-interval-mapping (MIM) models constructed with QGene 4.3 (Joehanes and Nelson 2008). The F statistics for main and interaction effects were collected from SAS output with Perl scripts, converted to LOD scores, and plotted against the corresponding QTL positions. With only two locations, the Q × L effect selected is that of location AR relative to LA. The interval-mapping model was also re-fitted for SB with the inclusion of DTH and HT as covariates with the aim of accounting for their influence on SB.
Model (1) was re-fitted with SAS PROC GLM with the Q terms replaced by G (i.e., DHL) so that approximate fractions of genetic variance explained could be computed for each of the QTLs as SS(Q)/SS(G), where SS denotes the Type III sum of squares and Q and G denote, respectively the main QTL or DHL effects.
For each of the three traits, the four or five QTLs accepted as showing main effects were combined in a single fit of model (1). The proportion of genetic variance accounted for by QTL main effects was calculated as \( [\sum\nolimits_{q} {\text{SS}} (Q_{q} )]/{\text{SS(}}G ) \) and the proportion of genetic × environment interaction (GEI) variation by Q × L as \( [\sum\nolimits_{q} {{\text{SS}}(Q_{q} *L)} ]/{\text{SS}}(GEI) \), where the q subscript indexes all QTLs included in the model, and GEI includes DHL × location, DHL × year, and DHL × year × location interaction terms.
Permutation testing to establish an acceptance threshold for Type I error control was carried out as follows: the original data set for the target trait was shuffled 500 times within strata; that is, all phenotypic values for a given DHL were exchanged as a block so that correlations between environments were preserved. For each shuffled data set, at least 100 QTL positions were randomly selected for model fitting, and the maximum F value for a chosen main or interaction effect was recorded, followed by ordering and selection of the 95th percentile value as an α = 0.05 cutoff.
Two-way interaction and permutation analysis were conducted for the field SB data as described in Nelson et al. (2011).
Comparison of QTL locations with those reported in the literature as lying on the same chromosome but near markers not common to both studies was done with reference to physical positions reported in Gramene for flanking markers in the respective genetic maps.
Results
Map, marker, and trait properties
Genetic map
The genetic map (Fig. 1) spanned approximately 1,350 of the 1,550 cM spanned by the rice reference map. Marker order was generally as expected from the rice genetic and physical maps (McCouch et al. 2002; Ware et al. 2002).

Genetic map produced from Cocodrie × MCR10277 doubled-haploid rice lines showing QTLs for sheath-blight disease, heading time, and plant height. Bars are centered on points of maximum LOD from multiple-interval mapping. Filled bars denote increasing effect from Cocodrie; open bars, from MCR10277. Trait abbreviations: SBF, SBI, SBM sheath-blight disease severity in field, microchamber, and mist chamber, respectively, DTH days to heading, HT plant height at maturity. Map modified from a plot by MapChart (Voorrips 2002)
Marker segregation
Regions of strong segregation distortion appeared on about half of chromosomes (Fig. 2), with those on chromosomes 2 and 8 containing several markers all with segregation skewed toward the same parent. Single markers showing large deviations from the expected 1:1 segregation were difficult to score reliably because of microsatellite peak multiplicity, but were retained as the best available polymorphic markers in the respective map regions. There was no consistent bias toward one parent and the overall genotype ratio was 10695:488:10434 (CC:CM:MM) or 49:2:48 with rounding. Of the unexpected (in DHLs) 2% heterozygosity, 94% of markers and 61% of DHLs showed at least one heterozygous locus.

Marker genotype segregation in Cocodrie × MCR10277 doubled-haploid rice lines. Numbered vertical divisions represent chromosomes on the genetic map and tick marks on x axis the positions of marker loci. y axis labels show proportions of the three non-missing genotypes, calculated at each marker. Solid line: MCR10277 homozygote; broken line: Cocodrie homozygote
Trait statistics
All three traits showed heritabilities close to 0.50 (Table 1) except for SB in the mist chamber. Cocodrie generally grew several cm taller than MCR10277, while heading up to 7 days earlier. The DHLs transgressed the parental means for all three traits (Fig. 3). Because the 2005 replicated trial in LA was intended only as a first survey of the DHLs, we chose not to include the data in the formal analyses, but the 2005 results were similar to those of 2006–2007 with respect to both field performance and QTL locations.

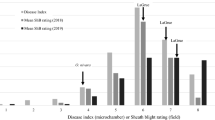
Phenotype distributions of three measured traits in Cocodrie × MCR10277 doubled-haploid rice lines in Crowley, LA in 2006. SB rating: field sheath blight severity, assessed on an 0–9 scale. Solid arrows indicate parent means
Correlations (Table 2) between days to heading and SB were high and negative in 2006 but lower in 2007, while plant height was negatively but weakly correlated with disease. All SB assays were generally well correlated, with r values (based on means across replications) ranging from 0.37 to 0.72. The parents were uniformly ranked in field and microchamber assays. However, in the mist chamber the correlations between replications ranged from 0.33 to 0.51. Here the DHLs showed more disease on average than the parents. Unexpectedly, Cocodrie showed less disease than MCR10277 in two of five trials and the two parents were grouped (results not shown) with five check cultivars including the resistant indica Jasmine 85 and separated from three checks including the susceptible Lemont, based on a Ryan–Einot–Gabriel–Welsch multiple-range test (SAS Institute 2004) applied to the parents and the eight checks.
QTL mapping results
Sheath blight
Four QTLs, all with the resistance allele from MCR, accounted for 47% of field genetic variation in the full-QTL models. A major QTL on chromosome 9, expressed in all assays and locations, reduced disease rating in the MCR10277 homozygote by up to 1.2 disease-severity units and accounted for 15–37% of genetic variation (Table 3; Fig. 4). In the field data, pronounced resistance effects were visible on chromosomes 6 (with resistance from MCR, but colocating with a major HT locus at which MCR contributed the increasing allele) and 8 (with resistance from Cocodrie, but colocating with a major DTH locus at which Cocodrie contributed the increasing allele). Since these disappeared when DTH and HT were included in the model, they were rejected as SBR QTLs. Three further SBR QTLs were identified on chromosomes 2 (two QTLs) and 12. All field-expressed QTLs appeared to confer at least some resistance in microchamber testing against the isolate RR0140-1 that was used for field inoculation in AR, even at loci where heading time and height showed influence on SBR. With respect to QTL × location interaction, for which a separate permutation threshold was computed, only qsbr_9.1 showed an effect, with the MCR allele tending to increase disease in AR. Year interaction effects fitted in the model were negligible. In the two-way QTL-interaction scan, no interactions approached the permutation threshold.

LOD-statistic and additive-effect profiles for full QTL models for three measured traits in Cocodrie × MCR10277 doubled-haploid rice lines in two field locations and one greenhouse (microchamber) assay. In all panels the upper profile shows the LOD for model fit and the lower profile the additive effect expressed in the units of measurement of the trait, while the black contour represents QTL main effect. Gray contour in all panels but c shows QTL × location effect. Horizontal dotted lines in the upper profiles of panels denote α = 0.05 significance thresholds established by permutation; vertical divisions demarcate chromosomes. a QTL profile for field SB, with model including year, location, and cofactor QTLs. b As a but with traits DTH and HT included as covariates. c Interval-mapping profiles for SB measured in the microchamber assay, with model including cofactor markers. d As a but with DTH as the response variable. e As a but with HT as the response variable
Days to heading
Four DTH QTLs, accounting for a total of 45% of genetic variation and each conferring from 2 to 3 days of DTH difference between homozygotes, were identified on chromosomes 5, 8, 9, and 12, with Cocodrie providing the increasing allele at the first three. SB resistance in the field was associated with the lateness alleles at the loci on chromosomes 8 and 12, but showed no association at the other two. Only qdth_8.1 showed a location interaction, but a minor one.
Plant height
Five height QTLs on chromosome 2, 3, 5, and 6 (with two) accounted for 62% of genetic variation. Despite the generally negative, though modest, correlation between SB disease and plant height (Table 2), the major chromosome-3 height QTL showed no association with SB (Fig. 4a, b) and the only map location at which inclusion of HT and DTH as covariates in the SB QTL model eliminated putative SBR QTLs was at qpht_6.2.
Discussion
Map coverage reflects limited polymorphism
The coverage gaps observed in Fig. 1 were not a property of the marker type or a product of inadequate polymorphism screening. As an example, the gap between RM1287 and RM1061 on chromosome 1, spanning 66 cM on the reference map, was screened with 27 microsatellite markers, with only one, adjacent to RM1287, polymorphic between the parents, and with a longest unsampled stretch of only 6.7 cM. In a separate study (Ware et al. 2002) of single-nucleotide polymorphisms (SNPs) typed in assorted rice cultivars, an additional 68 SNPs in the example chromosome-1 interval failed to distinguish Cocodrie from MCR10277 (unpublished data, W. Solomon et al.). This pattern of polymorphism islands was similar on other chromosomes. If the genetically informative distance on either side of a polymorphic marker is taken to be the lesser of 5 cM or half the distance to the next such marker, the total potentially informative distance in the population was 1,192 cM or 0.78 of the reference-map length, while the genotyped markers covered 0.79 of this distance. This assayed proportion increased to 0.86 if a window of 8 cM was allowed. Most missed QTLs are thus expected to be due to genomic similarity between the parents and not to inadequate marker coverage.
Apparent heterozygosity in doubled-haploid lines
The 2% heterozygosity rate in DHL marker genotypes is expected to be an artefact of genotype calling. In SSRs “stutter” confounds calls particularly when the alleles differ by only two nucleotides. Such less-preferred markers were retained when they were the only representatives of otherwise monomorphic map regions. Reassigning the apparent heterozygotes as missing data had no effect on the results.
Not all height or heading-time genes affect SB equally
Despite the negative correlation of disease with height in the DHLs, the taller, earlier parent Cocodrie is the more susceptible. That the major height allele at qpht_3.1, like the smaller qpht_5.1 and qpht_6.1, shows no statistical effect on disease suggests that these QTLs exert their effects on plant growth after infection has reached the canopy, while qpht_6.2 acts earlier enough in development to aid the rice plant in outgrowing the pathogen. QTL qpht_2.1 is a special case, as it coincides with a minor heading-time QTL such that the MCR alleles conferring earlier heading but taller stature appear to magnify SBR at locus qsbr_2.2.
As with height QTLs, heading-time QTLs also affect SBR differently. The three-day homozygote heading delay contributed by the Cocodrie allele at locus qdth_8.1 accounts for an 0.4-level drop in disease (not shown in Table 3 since this was not considered a SBR QTL). Likewise, resistance qsbr_12.1 lies at a DTH locus where the later-heading allele favors lower disease. However, qdth_5.1 and qdth_9.1, each with a 2-day heading difference contributed by Cocodrie, show no effect on SBR. These results suggest that it is not merely synchrony between plant and pathogen growth that influences resistance, but some more basic properties of plant developmental genes. Supporting this notion is the observation that even in the microchamber assay, which was completed well before plant heading, there are hints (Fig. 4c) that these developmental QTLs affect resistance to the same pathogen isolate in the same directions as seen in the field.
Relationship with QTLs in previous reports
Major SB QTLs lying in the chromosome-9 location of our qsbr_9.1 have been reported by previous authors: in 279 F2:3 families from a cross of tropical japonica cultivars Rosemont and Pecos (Sharma et al. 2009); in crosses of the resistant indica cultivar Jasmine 85 with the susceptible japonica Lemont (Liu et al. 2009; Zou et al. 2000); and in introgression lines from Lemont × Teqing (Pinson et al. 2005) tested with a U. S. isolate. Teqing is a highly resistant Chinese indica cultivar and, based on this finding, a possible source of the qsbr_9.1 resistance allele in the pedigree of MCR10277.
While genotype data for the pedigrees of these parents are insufficient for exhaustively tracing the origin of the resistance alleles, available data indicate both isolate specificity and diverse origins of the QTLs we have identified. At all 19 tested SSRs (as well as 52 SNPs) over the bottom 50 cM of chromosome 9 surrounding qsbr_9.1 the Teqing and MCR10277 alleles are identical, supporting the Teqing origin of the resistance allele. However, in reciprocal Lemont × Teqing introgression lines tested with a Chinese isolate during two years in the field, no QTL was identified in this region even at a very liberal P value threshold; of QTLs reported on chromosomes 4, 6, 7, 8, 10, and 11, each reduced disease by only around 1% and only the chromosome-4 QTL was expressed in both genetic backgrounds (Xie et al. 2008). It is also noteworthy that the 15 SSRs genotyped in the 45-cM terminal portion of chromosome 9 in resistant Pecos do not show the Teqing (and MCR10277) allele. The Pecos resistance at this locus thus appears to be from a different source and may represent a different QTL allele, a supposition consistent with the authors’ proposal that it originates in a japonica background. Comparison of the MCR10277 with the Jasmine SSR and SNP data for this map region shows a mixture of poly- and monomorphic loci, leaving uncertain the ancestral relationship of the QTL of Liu et al. (2009) and Zou et al. (2000) with our major QTL qsbr_9.1.
At the position of our qsbr_2.1, Liu et al. (2009) and Zou et al. (2000) both reported a Jasmine SB QTL; however, the first of these identified it in a mist-chamber assay using the same conditions and isolate used in our study, whereas it was not expressed in the mist chamber in our study. The QTL does not lie near those reported on chromosome 2 by other authors (Pinson et al. 2005; Sharma et al. 2009) and neighboring loci do not carry Teqing alleles. Similarly, qsbr_12.1 lies in a 16-cM stretch of chromosome 12 in which none of the 9 SSRs carried the Teqing allele. In contrast, qsbr_2.2 is centered in a 37-cM stretch of 13 SSRs all carrying Teqing alleles, yet was not expressed against the U. S. SB isolate used in the Lemont × Teqing cross of Li et al. (1995) even though our parental genotyping study shows ample polymorphism in that map region in that cross.
An F2:3 design such as that of Sharma et al. (2009) allows the estimation of dominance effects. Though these authors did not report such an analysis, reanalysis of their data (results not shown) confirms that both their chromosome-2 (from short-statured susceptible parent Rosemont) and chromosome-9 (from tall, resistant Pecos) QTLs confer dominant resistance.
SB isolate specificity
SBR shows some specificity with respect to pathogen isolate or plant growth stage, as reported also by Liu et al. (2009). SB disease produced by the Colombian isolate in 50-day-old plants was moderated mostly by qsbr_9.1, while that from both U. S. isolates provoked responses in both seedling and field-grown adult plants. The U. S. isolate was selected as a slower-growing one producing in microchamber assays disease levels better correlated to those in the field (Wamishe et al. 2007) than did more aggressive selections. The absence of correspondence of our SBR QTLs with those reported by Xie et al. (2008) in Teqing, an ancestor of MCR10277 showing good resistance to U. S. isolates, under challenge with a virulent Chinese SB isolate cautions against the hope of finding universally effective SBR genes. A challenge of the DHL set with multiple R. solani isolates would better elucidate isolate × cultivar interaction for this disease.
Correspondence of QTL numbers and effects with parental differences
If all QTL effects are accounted for in a mapping population and allele frequencies are near expected values, the sum of the additive effects in a multiple-QTL model should approximate half the difference between the parental means for the respective trait. This relationship held for SB, with the alleles at the four SBR loci conferring an effect of 1.7 disease levels, about half the mean difference between Cocodrie and MCR. However for DTH, the QTL effects totaled 1.4 days in favor of Cocodrie, whereas the observed 6.8-day delay of MCR predicted a sum of −3.4, and for HT the QTL effects totaled −2.5 cm favoring MCR, whereas Cocodrie’s height advantage predicted a value of 1.6. Despite the high proportion of genetic variation explained by the QTL models for these traits and a marker coverage as dense as allowed by parental polymorphism, some substantial QTLs or cryptic interactions remain unexplained.
Conclusions
The superior resistance of rice parent MCR10277 to sheath blight in contrast to susceptible Cocodrie is well supported by the identification of four MCR-contributed resistance QTLs identified in this study. The correspondence of SB QTLs detected in microchamber assays with those in the field encourages the use of this fast and easily replicated assay for practical resistance breeding. Beyond our scope but of future research interest is the question: how do plant developmental genes loosely classified as governing height or flowering time influence disease development differently? Analysis of a wider array of environments, ideally exploiting historical variety testing records in an association-mapping scheme, could supplement the use of controlled mapping populations for addressing this question. Though resistance genes with global effectiveness against sheath blight should not be expected, the SBR QTLs from MCR10277, particularly the chromosome-9 QTL, offer promise as a breeding target for partial resistance in southern U. S. and Latin American rice cultivars and a starting point for gene isolation.
Abbreviations
- DHL:
-
Doubled-haploid line
- SB:
-
Sheath blight
- QTL:
-
Quantitative-trait locus
References
Brooks S (2007) Sensitivity to a phytotoxin from Rhizoctonia solani correlates with sheath blight susceptibility in rice. Phytopathology 97:1207–1212
Channamallikarjuna V, Sonah H, Prasad M, Rao G, Chand S, Upreti H, Singh N, Sharma T (2010) Identification of major quantitative trait loci qSBR11–1 for sheath blight resistance in rice. Mol Breed 25:155–166
Che KP, Zhan QC, Xing QH, Wang ZP, Jin DM, He DJ, Wang B (2003) Tagging and mapping of rice sheath blight resistance gene. Theor Appl Genet 106:293–297
Chu QR, Linscombe SD, Rush MC, Groth DE, Oard J, Sha X, Utomo HS (2006) Registration of a C/M doubled haploid mapping population of rice. Crop Sci 46:1416
de Givry S, Bouchez M, Chabrier P, Milan D, Schiex T (2005) CarthaGene: multipopulation integrated genetic and radiation hybrid mapping. Bioinformatics 21:1703–1704
Dudley JW, Moll RH (1969) Interpretation and use of estimates of heritability and genetic variances in plant breeding. Crop Sci 9:257–262
Groth DE (2008) Effects of cultivar resistance and single fungicide application on rice sheath blight, yield, and quality. Crop Prot 27:1125–1130
Groth DE, Rush MC, Hollier CA (1991) Rice diseases and disorders in Louisiana Bull No 828. Louisiana Agricultural Experiment Station, Baton Rouge
Jia Y, Correa-Victoria F, McClung A, Zhu L, Liu G, Wamishe Y, Xie J, Marchetti MA, Pinson SRM, Rutger JN, Correll JC (2007) Rapid determination of rice cultivar response to the sheath blight pathogen Rhizoctonia solani using a micro-chamber screening method. Plant Dis 91:485–489
Jiang C, Zeng Z-B (1997) Mapping quantitative trait loci with dominant and missing markers in various crosses from two inbred lines. Genetica 101:47–58
Joehanes R, Nelson JC (2008) QGene 4.0, an extensible Java QTL-analysis platform. Bioinformatics 24:2788–2789
Lee FN, Rush MC (1983) Rice sheath blight: a major rice disease. Plant Dis 67:829–832
Li ZK, Pinson SRM, Marchetti MA, Stansel JW, Park WD (1995) Characterization of quantitative trait loci (QTLs) in cultivated rice contributing to field resistance to sheath blight (Rhizoctonia solani). Theor Appl Genet 91:382–388
Linscombe DS, Jodari F, Bollich PK, Groth DE, White LM, Chu QR, Dunand RT, Sanders DE (2000) Registration of ‘Cocodrie’ rice. Crop Sci 40:294
Liu G, Jia Y, Correa-Victoria F, Prado G, Yeater K, McClung A (2009) Mapping quantitative trait loci responsible for resistance to sheath blight in rice. Phytopathology 99:1078–1084
Mackill DJ, McKenzie KS (2003) Origin and characteristics of U. S. rice cultivars. In: Smith CW, Dilday RH (eds) Rice: origin history technology, and production. John Wiley & Sons, Hoboken, pp 87–100
Marchetti MA, Bollich CN (1991) Quantification of the relationship between sheath blight severity and yield loss in rice. Plant Dis 75:773–775
McCouch SR, Teytelman L, Xu YB, Lobos KB, Clare K, Walton M, Fu BY, Maghirang R, Li ZK, Xing YZ, Zhang QF, Kono I, Yano M, Fjellstrom R, DeClerck G, Schneider D, Cartinhour S, Ware D, Stein L (2002) Development and mapping of 2240 new SSR markers for rice (Oryza sativa L.). DNA Res 9:199–207
Nelson JC, McClung AM, Fjellstrom RG, Moldenhauer KAK, Boza E, Jodari F, Oard JH, Linscombe S, Scheffler BE, Yeater KM, Guo Z (2011) Mapping QTL main and interaction influences on milling quality in elite U.S. rice germplasm. Theor Appl Genet 122:291–309
Pan XB, Rush MC, Sha XY, Xie QJ, Linscombe SD, Stetina SR, Oard JH (1999) Major gene, nonallelic sheath blight resistance from the rice cultivars Jasmine 85 and Teqing. Crop Sci 39:338–346
Pinson SRM, Capdevielle FM, Oard JH (2005) Confirming QTLs and finding additional loci conditioning sheath blight resistance in rice using recombinant inbred lines. Crop Sci 45:503–510
Richards CM, Reilley A, Touchell D, Antolin MF, Walters C (2004) Microsatellite primers for Texas wild rice (Zizania texana), and a preliminary test of the impact of cryogenic storage on allele frequency at these loci. Conserv Genet 5:853–859
Rodrigues FÁ, Datnoff LE, Korndörfer GH, Seebold KW, Rush MC (2001) Effect of silicon and host resistance on sheath blight development in rice. Plant Dis 85:827–832
Rodrigues FÁ, Vale FXR, Datnoff LE, Prabhu AS, Korndörfer GH (2003) Effect of rice growth stages and silicon on sheath blight development. Phytopathology 93:256–261
SAS Institute (2004) SAS 9.1.3 help and documentation. SAS Institute, Inc., Cary
Sharma A, McClung AM, Pinson SRM, Kepiro JL, Shank AR, Tabien RE, Fjellstrom R (2009) Genetic mapping of sheath blight resistance QTLs within tropical japonica rice cultivars. Crop Sci 49:256–264
Slaton NA, Cartwright RD, Meng J, Gbur EE, Norman RJ (2003) Sheath blight severity and rice yield as affected by nitrogen fertilizer rate, application method, and fungicide. Agron J 95:1489–1496
Voorrips RE (2002) MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered 93:77–78
Wamishe YA, Jia Y, Singh P, Cartwright RD (2007) Identification of field isolates of Rhizoctonia solani to detect quantitative resistance in rice under greenhouse conditions. Front Agric China 1:361–367
Ware DH, Jaiswal PJ, Ni JJ, Yap I, Pan XK, Clark KY, Teytelman L, Schmidt SC, Zhao W, Chang K, Cartinhour S, Stein LD, McCouch SR (2002) Gramene, a tool for grass genomics. Plant Physiol 130:1606–1613
Webster RK, Gunnell PS (1992) Compendium of rice diseases. American Phytopathological Society, St. Paul
Xie QJ, Linscombe SD, Rush MC, Jodari KF (1992) Registration of LSBR-33 and LSBR-5 sheath blight resistant germplasm lines of rice. Crop Sci 32:507
Xie X, Xu M, Zang J, Sun Y, Zhu L, Xu J (2008) Genetic background and environmental effects on expression of QTL for sheath blight resistance in reciprocal introgression lines of rice. Acta Agron Sin 34:1885–1893
Yin Y, Zuo S, Wang H, Chen Z, Gu S, Zhang Y, Pan X (2009) Evaluation of the effect of qSB-9Tq involved in quantitative resistance to rice sheath blight using near-isogenic lines. Can J Plant Sci 89:731–737
Zou JH, Pan XB, Chen ZX, Xu JY, Lu JF, Zhai WX, Zhu LH (2000) Mapping quantitative trait loci controlling sheath blight resistance in two rice cultivars (Oryza sativa L.). Theor Appl Genet 101:569–573
Acknowledgments
Support for this study was provided by U. S. Department of Agriculture Cooperative State Research, Education and Extension Service–National Research Initiative–Applied Plant Genomics Program grant 2004-35317-14867 entitled “RiceCAP: A coordinated research, education, and extension project for the application of genomic discoveries to improve rice in the United States.” This is contribution 10-308-J from the Kansas Agriculture Experiment Station.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nelson, J.C., Oard, J.H., Groth, D. et al. Sheath-blight resistance QTLS in japonica rice germplasm. Euphytica 184, 23–34 (2012). https://doi.org/10.1007/s10681-011-0475-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10681-011-0475-1