
Abstract
Multiplex PCR is practically a reasonable choice for molecular marker-assisted selection in potato breeding. We had developed and were using a multiplex PCR method for selection of resistance genes to cyst nematode (H1), Potato virus X (Rx1) and late blight (R1 and R2). Since then, more reliable and tightly linked markers for H1 and R2, and a new marker for resistance to Potato virus Y (Ry chc ) were developed. In this article, all these superior markers, including a positive marker to eliminate PCR-failed samples, were incorporated into one multiplex PCR assay. Using the newly developed multiplex PCR technique, five plants potentially harboring all five resistance genes were selected from 96 hybrid plants approximately 5 h after DNA extraction, which is a third of the operation time compared with separate PCR reactions for each marker.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Conferring disease and pest resistances is important in crop breeding. Biological assays for resistance in a greenhouse or field are fundamental, but time- and space-consuming. Alternatively, DNA marker-assisted selection (MAS) can be used without special facilities for respective biological assays and not influenced by growth stages or growing conditions such as temperature, humidity, light intensity, day-length, etc. MAS can identify rapidly and reliably resistant genotypes, becoming an important and practical breeding tool (Babu et al. 2004; Xu and Crouch 2008). Replacement of genetic background by backcrosses to cultivated germplasm without losing desirable genes from wild materials can be facilitated using MAS technology (Tanksley et al. 1989; Barone 2004).
In the potato breeding program of Nagasaki Agricultural and Forestry Technical Development Center, Japan, DNA markers have been used to select genotypes with multiple resistances to potato cyst nematodes, Potato Virus Y (PVY), Potato virus X (PVX), and potato late blight. Of potato cyst nematodes [Globodera rostochiensis (Woll.) Behrens], Ro1 is only pathotype found in Japan and has increasingly become a serious problem particularly in seed potato production areas where once cyst nematodes are found, seed potatoes are not allowed to transport outside the areas (Mori et al. 2007). A single dominant gene H1, introduced from the accession CPC1673 of Solanum tuberosum L. ssp. andigena Hawkes, confers perfect resistance to the pathotype Ro1 (Huijsman 1955). Harboring H1 is now a prerequisite to the release of new cultivars in Japan. H1 was mapped to potato chromosome 5 (Pineda et al. 1993; Gebhardt et al. 1993). The high-resolution map of H1 provided tightly linked AFLP markers (Bakker et al. 2004), from which Ohbayashi et al. (2010) developed a sequence tagged site (STS) marker PCN. The recombination frequency between PCN and H1 is 7.4%. Recently, a set of more tightly linked markers N146 and N195 were developed, which sandwich H1 with the recombination frequencies 0.109 and 0.207%, respectively (Takeuchi et al. 2008; personal communication).
Twelve viruses are known to infect and damage potatoes in Japan (Maoka et al. 2010). Among them, PVY is the most important, and thus, resistance to PVY is highly desirable. Extreme resistance genes to PVY have been known from three different sources: Ry sto from S. stoloniferum Schlechtd. et Bché. (Cockerham 1943), Ry adg from S. tuberosum ssp. andigena (Munoz et al. 1975) and Ry chc from S. chacoense Bitt. (Asama et al. 1982). Ry chc was identified in one of Japanese leading cultivars, ‘Konafubuki’ (Hosaka et al. 2001), and mapped to the most distal end of chromosome 9 (Sato et al. 2006). The Ry chc -linked RAPD marker 38-530 has been used for MAS, although the recombination frequency was relatively high, 16.3% in the tetraploid population (Hosaka et al. 2001) and 0.9% in the diploid population (Sato et al. 2006). Recently, Takeuchi et al. (2008; personal communication) developed an STS marker Ry186, tightly linked with a recombination frequency of 0.203%.
The second important virus is PVX, which causes heavier mosaic symptoms if the plants are co-infected with PVY. A dominant resistance gene Rx1, or most likely Rx adg , incorporated from S. tuberosum ssp. andigena, and Rx2, or most likely Rx acl , incorporated from S. acaule Bitt., were mapped to chromosome 12 and 5, respectively (Ritter et al. 1991), and both were cloned (Bendahmane et al. 1999, 2000). Based on the sequence information of Rx (Bendahmane et al. 1999), Ohbayashi et al. (2010) developed an STS marker PVX, linked to Rx1 with the recombination frequency of 1.3%.
Potato late blight caused by Phytophthora infestans (Mont.) de Bary is the most important fungus disease of potatoes worldwide. Eleven hypersensitivity-type resistance genes (R genes) have been introgressed into potato cultivars from a Mexican hexaploid wild species S. demissum Lindl. (Black et al. 1953; Ross 1986). R1 is located on potato chromosome 5 (Leonards-Schippers et al. 1992), which was cloned and the R1-specific primers have been published (Ballvora et al. 2002). However, introgression of R genes conferring race-specific resistance into potato cultivars provided only transient resistance to late blight, as new races rapidly overcome the R gene-mediated resistance (Fry et al. 1993). Indeed, R1 is no more effective in Japan. Interestingly, the consistent quantitative trait locus (QTL) for resistance to late blight was mapped to the same location of R1 in populations originating from numerous Solanum species, using various races of P. infestans, and different resistance testing methods (Simko 2002). This supports Gebhardt’s hypothesis that qualitative and quantitative resistance phenotypes to P. infestans might be the product of different alleles of the same genetic locus and the hypersensitive cell death triggered by the R1 allele would be the extreme expression of a quantitative defense response induced by variants of the same gene product (Gebhardt 1994). Kuang et al. (2005) found that R1 is actually a resistance gene cluster. Thus, MAS for R1 may still be useful, although allele specific markers for the broader spectrum QTL resistance would be needed.
The R2 allele conferring resistance to P. infestans has been mapped to potato chromosome 4 (Li et al. 1998). In Japan, R2 was introgressed into ‘Hokkai 56’, which is a superior parental line for breeding late blight resistant cultivars. Based on the sequence information of AFLP markers tightly linked to R2 (Li et al. 1998), Ohbayashi et al. (2010) developed an STS marker R2-974 using a segregating population from ‘Hokkai 56’ (resistant) × ‘Norchip’ (susceptible). Later, it was found that R2-974 was missing in ‘Saya-akane’ that also has R2 introgressed from a completely different source material. Further improvement using a segregating population from ‘Saya-akane’ (resistant) × ‘Norin 1’ (susceptible) generated another STS marker R2-800, which could discriminate all R2 holders from the others (Ohbayashi et al. 2010).
We had assayed using these markers separately for all samples, which linearly increased the number of polymerase chain reaction (PCR), operation time and cost as the number of tested markers increased. To integrate MAS as a practical tool into a breeding program, reducing cost and increasing scale and efficiency are crucial (Xu and Crouch 2008). The first example for using PCR-based markers to select potato clones with multiple, monogenic resistance traits was reported by Gebhardt et al. (2006). 110 F1 plants were first screened with SCAR marker RYSC3 for PVY resistance, then positive ones tested with Gro1-4 for nematode resistance, and then double-positive ones tested with CAPS marker CP60 for PVX resistance. Such consecutive screening significantly reduced time and cost (Gebhardt et al. 2006). Alternatively, multiplex PCR with several diagnostic markers mixed together in one PCR reaction is preferable to reduce the number of PCR reactions. Multiplex PCR methods were widely used in detecting genetically modified crops (de la Cerda et al. 2009; Randhawa et al. 2009), identifying the allele composition of high-molecular weight glutenin complex loci in wheat (Moczulski and Salmanowicz 2003), or detecting respiratory viruses (Mahony et al. 2009), and the low-cost, high-throughput performance has been demonstrated (Frey et al. 2004; Mahony et al. 2009; Utomo et al. 2009).
Previously, we developed a multiplex PCR method to detect H1, Rx1, R1, and R2 genes using markers, PCN, PVX, R1, and R2-974, respectively (Mori et al. 2010). In a course of screening, we found that ‘Hokkai 56’, used frequently as an R2 resistance source, was judged H1 positive by using the PCN marker, although it actually does not have H1. In addition, Ry chc had been assayed separately by RAPD marker 38-530, because it was unable to be included into the multiplex PCR due to a very different annealing temperature. Recent development of more tightly linked, specific STS markers rendered us to improve our multiplex PCR method. Because these new markers have demonstrated perfect coincidence with resistance phenotypes in many cultivars and breeding lines of various levels of relatedness (Takeuchi et al. 2008; Ohbayashi et al. 2010) and the simultaneous detection method will provide wide applicability in various breeding programs. In this article, we report the improvement process of the current multiplex PCR method by replacing PCN by a set of flanking markers N146 and N195, R2-974 by R2-800, and adding Ry186. Further improvement is an addition of the granule-bound starch synthase I gene (GBSS) marker which is shared in all potato species, thus, it functions as a positive control to check whether the PCR was performed correctly or not. The newly developed multiplex PCR method significantly reduced operation time.
Materials and methods
Plant materials
A Japanese leading variety ‘Konafubuki’, a breeding line ‘T07137-6’, their hybrid family ‘T09068’ (96 genotypes), three other varieties (‘Dejima’, ‘Nishiyutaka’, and ‘Aiyutaka’) and three breeding lines (‘Hokkai 56’, ‘Saikai 35’, and ‘T08043-32’) were used in this study. Resistance genes of parental lines and varieties are shown in Table 1.
DNA extraction and DNA marker detection method
Total DNA was extracted from a piece of fresh leaf (approximately 1 cm2) using an automatic DNA extraction system (a combination of SH-48, CS-16, and PI-80X, KURABO, Japan) following the manufacturer’s instruction. DNA concentration (3–83 ng/μl) was measured using DyNA Quant™ 200 (Hoefer Pharmacia Biotech Inc., CA, USA).
The presence or absence of disease resistance genes was estimated by the following diagnostic DNA markers: a set of N146 and N195 for H1 (Takeuchi et al. 2008, personal communication), PVX for Rx1 (Ohbayashi et al. 2010), R1 for R1 (Ballvora et al. 2002), R2-800 for R2 (Ohbayashi et al. 2010), and Ry186 for Ry chc (Takeuchi et al. 2008, personal communication). As a positive control, PCR marker for the GBSS (Takeuchi et al. 2008, personal communication) was also used. Primer sequences and optimized concentration and annealing temperature for each PCR marker are shown in Table 2.
For marker detection using individual PCRs, the total volume was 10 μl consisting of 2 μl of template DNA, 5 μl of Ampdirect® Plus (Shimadzu Co., Japan), 0.25 units Taq DNA polymerase (Nova Taq ™ Hot Start DNA Polymerase, Novagen®, USA) and a set of primers (Table 2). Thermal cycling was performed using Veriti® 96-well thermal cycler (Applied Biosystems) (one cycle of 10 min at 94°C, followed by 35 cycles of 30 s at 94°C, 30 s at an annealing temperature described in Table 2, and 1 min, or 1.5 min only for R1, at 72°C, and then, terminated with one cycle of 5 min at 72°C). PCR products were separated by electrophoresis on a 1.4% agarose gel.
To optimize the multiplex PCR conditions, concentrations of primer sets, the amount of Taq DNA polymerase and the thermal profile for PCR reactions were varied. The optimized concentration of each primer set for multiplex PCR is shown in Table 2. All seven primer sets were mixed together into one reaction. The amount of Taq DNA polymerase was increased to 0.5 units. The other components in a reaction were the same as those of individual PCR. Thermal cycling was done by one cycle of 10 min at 94°C, five cycles of 30 s at 94°C, 30 s at 68°C, and 1.5 min at 72°C, 35 cycles of 30 s at 94°C, 30 s at 58°C and 1.5 min at 72°C, and then, terminated with one cycle of 5 min at 72°C.
Results
DNA concentration and evaluation by individual PCRs
DNA samples extracted by the automatic DNA extraction system were of the concentration range from 3 to 83 ng/μl with an average of 23.6 ng/μl (SD = 16.9). These were used as template DNA in PCR without adjusting DNA concentration. Presence or absence of marker bands in cultivars and breeding lines were assayed by individual PCRs, which perfectly matched with those of reported resistance phenotypes (Table 1); that is, ‘Nishiyutaka’ does not have any resistance genes, while the others have some of them. A cyst nematode susceptible cultivar ‘Hokkai 56’ was expectedly negative for N146 and N195 markers, which had been falsely positive by the previously used marker PCN (Mori et al. 2010).
Development of multiplex PCR
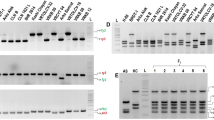
Using eight cultivars and breeding lines, concentrations of PVX and R1 primer sets were varied and the best concentration combination was determined (1.50 and 0.75 μM, respectively). Then, the next primer set of R2-800 was added and varied to different concentrations. Such a procedure of step by step addition and determination of the best concentration combination was performed in order of PVX, R1, R2-800, Ry186, N146, N195, and GBSS (Fig. 1). Annealing temperature was fixed to 58°C throughout the experiments. The presence or absence of expected marker bands (Table 1) and the band intensities were criteria for optimization. As shown in Fig. 1, when the concentration of a newly added primer set was too low, the band intensity became faint, while when it was too high, the band intensity increased and unspecific faint bands sometimes appeared (see unspecific faint bands of N195 at 0.10 μM concentration at step 5). As a result, after the determination of 1.50 and 0.75 μM combination for PVX and R1, respectively, 0.25 μM for R2-800, 0.10 μM for Ry186, 0.05 μM for N146, 0.05 μM for N195, and finally 0.15 μM for a positive control marker GBSS were determined. At this moment, the band intensities of all marker bands seemed a little faint. We increased the amount of Taq DNA polymerase from 0.25 to 0.5 units (step 7), which greatly improved the intensities of the marker bands (Fig. 1).

Optimization by varying the concentration of newly added primers, and finally adding double dosage of Taq DNA polymerase (optimized ones in Bold). 1 ‘Dejima’, 2 ‘Aiyutaka’, 3 ‘Hokkai 56’, 4 ‘Saikai 35’, 5 ‘Konafubuki’, 6 ‘T07137-6’, 7 ‘T08043-32’, 8 ‘Nishiyutaka’, M lambda DNA HindIII/EcoRI double-digests (NIPPON GENE Co., Ltd., Tokyo)
Detection sensitivity to different template DNA concentrations
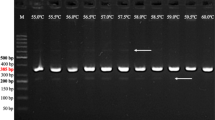
One of the hybrid genotypes ‘T09068-57’, which had all seven marker bands, and ‘Nishiyutaka’, which lacked all but a positive control GBSS marker band, were examined by the multiplex PCR method described above for a series of template DNA concentrations (15, 10, 5, 2.5, 2, 1.5, 1, 0.5, and 0.1 ng/μl of PCR reaction volume). In ‘T09068-57’, all marker bands were detected at 2.5 ng/μl or higher concentrations, while at less than 2.5 ng/μl concentrations PVX and R1 marker bands were not detectable (Fig. 2a). In ‘Nishiyutaka’, any marker bands or unspecific bands were not detected except for the positive control GBSS, which was detected at the concentration as low as 0.5 ng/μl.

Effects of template DNA concentrations (ng/μl of PCR reaction volume) and thermal profiles. Annealing temperature constant at 58°C (a) or touchdown from 68 to 58°C (b). ‘T09068-57’ has all five resistance gene markers, while ‘Nishiyutaka’ has none. M 100 bp DNA ladder (0.1–1.0 and 1.5 kbp) (NIPPON GENE Co., Ltd., Tokyo)
Further improvement of detection sensitivity
The positive control GBSS marker was detected even at the template DNA concentration of 0.5 or 0.1 ng/μl, so that there is a possibility to falsely judge PVX and R1 markers as negative because the latter two markers were difficult to detect at the concentration less than 2.5 ng/μl (Fig. 2a). In order to improve detection sensitivity of PVX and R1, the concentration of R1 primer set was reduced from 0.75 to 0.25 μM, because PVX and R1 markers were likely in a compensatory relation in PCR, where lowering the relative amount of R1 primer set increased the band intensity of PVX (Mori et al. 2010). Furthermore, five cycles of 30 s at 94°C, 30 s at 68°C, and 1.5 min at 72°C were added prior to the normal cycles (touchdown PCR), which supposedly amplify selectively R1 marker because the only R1 had such a high annealing temperature (Table 2). Consequently, PVX and R1 markers were successfully detected at the concentration as low as 0.5 ng/μl (Fig. 2b).
Evaluation and segregation of the hybrid genotypes
Ninety-six genotypes of ‘T09068’ family from a cross between ‘Konafubuki’ and ‘T07137-6’ were evaluated by the improved multiplex PCR method, the results of which perfectly coincided with those produced using the individual PCR method (Fig. 3). Marker bands were detected in either one of parents, so that all but R1 segregated in a simplex fashion in the hybrid progeny (Table 3). As expected, N146 and N195 co-segregated perfectly. Segregation ratios of N146, N195, and R2-800 markers fit more to the random chromatid assortment model, while Ry186 and PVX markers fit more to the random chromosome assortment model. The R1 marker was over-represented, indicating that the parental line ‘T07137-6’ was likely duplex for this gene. The segregation ratio of the R1 marker fitted only the random assortment model with relatively low probability. Although both H1 and R1 map to chromosome 5, 55 genotypes had either H1 or R1 marker. Among 96 genotypes five had all marker bands. Conventional biological assays confirmed their resistances at least for those available to be assayed at the time (data not shown).

Evaluation of the hybrid family T09068 by the improved multiplex PCR method. M 100 bp DNA ladder (0.1–1.0 and 1.5 kbp) (NIPPON GENE Co., Ltd., Tokyo)
Comparison of operation time
Ninety-six genotypes of ‘T09068’ family were evaluated for each marker using the individual PCR method. Only positive samples were subjected to the next marker evaluation. Consequently, the cumulative sample number was 182, and the total time consumed was 997 min (Table 4). In contrast, evaluation using the multiplex PCR method consumed 308 min (significant reduction to 30.9%).
Discussion
We successfully developed the multiplex PCR method, which could detect simultaneously the DNA markers for resistance genes, H1, Rx1, R1, R2, and Ry chc . This method is superior to the previous one (Mori et al. 2010), because (1) H1 can be detected by more tightly linked markers sandwiching H1, (2) Ry chc can now be assayed by a tightly linked marker, (3) R2 can be more accurately distinguished, and (4) false negatives can be avoided by inclusion of GBSS marker.
Time and cost are major issues in adopting MAS to practical breeding programs (Xu and Crouch 2008; Mahony et al. 2009; Utomo et al. 2009). As compared with the marker by marker selection (Gebhardt et al. 2006), the newly developed multiplex PCR method reduced to 52.7% in the number of PCR reactions and to 30.9% in the time consumed. Accordingly, although the cost of Taq DNA polymerase became doubled, the total cost reduced significantly. The automated DNA extraction system generated DNA of an average concentration of 23.6 ng/μl, and 2 μl from each sample were used in a volume of 10 μl PCR reaction (=4.72 ng/μl), which was far over the sensitivity limit as low as 0.5 ng/μl. The lowest DNA concentration of a sample was 3 ng/μl (=0.6 ng/μl in a PCR reaction), which was still over the sensitivity limit. Therefore, the adjustment of DNA concentration prior to PCR is not needed, which surely facilitates adopting MAS into practical breeding programs.
Based on the segregation ratio of R1 marker in a tetraploid hybrid population ‘T09068’ (from ‘Konafubuki’ × ‘T07137-6’), the parental line ‘T07137-6’ could be duplex for R1 (R1R1r1r1). Since both parents of ‘T07137-6’ (‘Hokkai 56’ and ‘Aiyutaka’) harbor R1 (Table 1), the duplex condition of R1 in ‘T07137-6’ is understandable. Both H1 and R1 map to chromosome 5, but the progeny did not show co-segregation between the H1 and R1 markers. Given this and the segregation ratios (Table 3) it is likely that the parent ‘T07137-6’ had two homologous chromosomes harboring the R1 gene and another harboring the H1 gene.
In conclusion, we developed the new multiplex PCR method, by which from 96 inter-varietal hybrid genotypes we could identify five genotypes having all five resistance gene markers with reduced time and cost. To our best knowledge, this is the most practical MAS application in potato breeding. We are now using it as a practical tool in breeding varieties with multiple resistances.
References
Asama K, Ito H, Murakami N, Itoh T (1982) New potato variety “Konafubuki”. Bull Hokkaido Pref Agric Exp Stn 48:75–84
Babu R, Nair SK, Prasanna BM, Gupta HS (2004) Integrating marker-assisted selection in crop breeding-prospects and challenges. Cur Sci 87:607–619
Bakker E, Achenbach U, Bakker J, van Vliet J, Peleman J, Segers B, van der Heijden S, van der Linde P, Graveland R, Hutten R, van Eck H, Coppoolse E, van der Vossen E, Bakker J, Goverse A (2004) A high-resolution map of the H1 locus harbouring resistance to the potato cyst nematode Globodera rostochiensis. Theor Appl Genet 109:146–152
Ballvora A, Ercolano MR, Weiß J, Meksem K, Bormann CA, Oberhagemann P, Salamini F, Gebhardt C (2002) The R1 gene for potato resistance to late blight (Phytophthora infestans) belongs to the leucine zipper/NBS/LRR class of resistance genes. Plant J 30:361–371
Barone A (2004) Molecular marker-assisted selection for potato breeding. Am J Pot Res 81:111–117
Bendahmane A, Kanyuka K, Baulcombe DC (1999) The Rx gene from potato controls separate virus resistance and cell death responses. Plant Cell 11:781–791
Bendahmane A, Querci M, Kanyuka K, Baulcombe DC (2000) Agrobacterium transient expression system as a tool for the isolation of disease resistance genes: application to the Rx2 locus in potato. Plant J 21:73–81
Black W, Mastenbroek C, Mills WR, Peterson LC (1953) A proposal for an international nomenclature of races of Phytophthora infestans and of genes controlling immunity in Solanum demissum derivatives. Euphytica 2:173–179
Cockerham G (1943) Potato breeding for virus resistance. Ann Appl Biol 30:105–108
de la Cerda CGM, Herrera RR, Gonzalez CNA, Valdés MHR (2009) Multiplex-PCR detection of accompanying GMO transgenic sequences. Int J Eng Techn 1:285–287
Frey JE, Frey B, Sauer C, Kellerhals M (2004) Efficient low-cost DNA extraction and multiplex fluorescent PCR method for marker-assisted selection in breeding. Plant Breed 123:554–557
Fry WE, Goodwin SB, Dyer AT, Matuszak JM, Drenth A, Tooley PW, Sujkowski LS, Koh YJ, Cohen BA, Spielman LJ, Deahl KL, Inglis DA, Sandlan KP (1993) Historical and recent migrations of Phytophthora infestans: chronology, pathways, and implications. Plant Dis 77:653–661
Gebhardt C (1994) RFLP mapping in potato of qualitative and quantitative genetic loci conferring resistance to potato pathogens. Am Potato J 71:339–345
Gebhardt C, Mugniery D, Ritter E, Salamini F, Bonnel E (1993) Identification of RFLP markers closely linked to the H1 gene conferring resistance to Globodera rostochiensis in potato. Theor Appl Genet 85:541–544
Gebhardt C, Bellin D, Henselewski H, Lehmann W, Schwarzfischer J, Valkonen JPT (2006) Marker-assisted combination of major genes for pathogen resistance in potato. Theor Appl Genet 112:1458–1464
Hosaka K, Hosaka Y, Mori M, Maida T, Matsunaga H (2001) Detection of a simplex RAPD marker linked to resistance to potato virus Y in a tetraploid potato. Am J Pot Res 78:191–196
Huijsman CA (1955) Breeding for resistance to the potato root eelworm. 2. Data on the inheritance of resistance in andigenum–tuberosum crosses obtained in 1954. Euphytica 4:133–140
Kuang H, Wei F, Marano MR, Wirtz U, Wang X, Liu J, Shum WP, Zaborsky J, Tallon LJ, Rensink W, Lobst S, Zhang P, Tornqvist CE, Tek A, Bamberg J, Helgeson J, Fry W, You F, Luo MC, Jiang J, Buell CR, Baker B (2005) The R1 resistance gene cluster contains three groups of independently evolving, type I R1 homologues and shows substantial structural variation among haplotypes of Solanum demissum. Plant J 44:37–51
Leonards-Schippers C, Gieffers W, Salamini F, Gebhardt C (1992) The R1 gene conferring race-specific resistance to Phytophthora infestans in potato is located on potato chromosome V. Mol Gen Genet 233:278–283
Li X, van Eck HJ, Rouppe van der Voort JNAM, Huigen DJ, Stam P, Jacobsen E (1998) Autotetraploids and genetic mapping using common AFLP markers: the R2 allele conferring resistance to Phytophthora infestans mapped on potato chromosome 4. Theor Appl Genet 96:1121–1128
Mahony JB, Blackhouse G, Babwah J, Smieja M, Buracond S, Chong S, Ciccotelli W, O’Shea T, Alnakhli D, Griffiths-Turner M, Goeree R (2009) Cost analysis of multiplex PCR testing for diagnosing respiratory virus infection. J Clin Microbiol 47:2812–2817
Maoka T, Sugiyama S, Maruta Y, Hataya T (2010) Application of cDNA microarray for simultaneous detection of 12 potato viruses. Plant Dis 94:1248–1254
Moczulski M, Salmanowicz BP (2003) Multiplex PCR identification of wheat HMW glutenin subunit genes by allele-specific markers. J Appl Genet 44:459–471
Mori K, Ohbayashi K, Tamiya S, Sakamoto Y, Mukojima N, Nakao T, Hosaka K (2010) Development of a simultaneous detection method of DNA markers linked to four disease and pest resistance genes in potato using multiplex PCR. Breed Res 12:22–25
Mori M, Tsuda S, Mukojima N, Kobayashi A, Matsuura-Endo C, Ohara-Takada A, Zaidul ISM (2007) Breeding of potato cyst nematode resistant varieties in Japan. In: Haverkort AJ, Anisimov BV (eds) Potato production and innovative technologies. Wageningen Academic Publishers, The Netherlands, pp 328–339
Munoz FJ, Plaisted RL, Thurston HD (1975) Resistance to potato virus Y in Solanum tuberosum ssp. andigena. Am Potato J 52:107–115
Ohbayashi K, Nakata N, Chaya M, Komura K (2010) Development of a detection method of resistance to potato disease and pest using DNA markers. 1. Detection methods of resistance to potato virus X, potato cyst nematode and late blight. Bull Nagasaki Agri Fore Technol Dev Cen 1:1–26
Pineda O, Bonierbale MW, Plaisted RL (1993) Identification of RFLP markers linked to the H1 gene conferring resistance to the potato cyst nematode Globodera rostochiensis. Genome 36:152–156
Randhawa GJ, Singh M, Sharma R (2009) Validation of ST-LS1 as an endogenous reference gene for detection of AmA1 and cry1Ab genes in genetically modified potatoes using multiplex and real time PCR. Am J Pot Res 86:398–405
Ritter E, Debener T, Barone A, Salamini F, Gebhardt C (1991) RFLP mapping on potato chromosomes of two genes controlling extreme resistance to potato virus X (PVX). Mol Gen Genet 227:81–85
Ross H (1986) Potato breeding-problems and perspectives. Verlag Paul Parey, Berlin
Sato M, Nishikawa K, Komura K, Hosaka K (2006) Potato virus Y resistance gene, Ry chc , mapped to the distal end of potato chromosome 9. Euphytica 149:367–372
Simko I (2002) Comparative analysis of quantitative trait loci for foliage resistance to Phytophthora infestans in tuber-bearing Solanum species. Am J Pot Res 79:125–132
Takeuchi T, Sasaki J, Suzuki T, Horita H, Iketani S (2008) High-resolution maps and DNA markers of the Potato virus Y resistance gene Ry chc and the potato cyst nematode resistance gene H1. Breed Res 10 (Suppl 1):148
Tanksley SD, Young ND, Paterson AH, Bonierbale MW (1989) RFLP mapping in plant breeding: new tools for an old science. BioTechnology 7:257–263
Utomo HS, Wenefrida I, Blanche SB, Linscombe SD (2009) Low-cost method for streamlining marker-assisted selection and breeding line development in rice (Oryza sativa L.). Int J Genet Mol Biol 1:64–74
Xu Y, Crouch JH (2008) Marker-assisted selection in plant breeding: from publications to practice. Crop Sci 48:391–407
Acknowledgments
This study was supported in part by the Ministry of Agriculture, Forestry and Fisheries of Japan (Rural Bio-Mass Research Project, BCD-11232). We highly appreciate Tetsuo Maoka and Takashi Narabu, National Agricultural Research Center for Hokkaido Region, Tetsuji Ogawa, Nagasaki Agricultural and Forestry Technical Development Center, and Mikiko Kawahara and Yasuhiro Ogawa, Nagasaki Pest Control Station, for performance of or assistance in biological assays of disease resistances.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mori, K., Sakamoto, Y., Mukojima, N. et al. Development of a multiplex PCR method for simultaneous detection of diagnostic DNA markers of five disease and pest resistance genes in potato. Euphytica 180, 347–355 (2011). https://doi.org/10.1007/s10681-011-0381-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10681-011-0381-6