
Abstract
A quantitative trait loci (QTL) associated with resistance to pea rust, caused by the fungus Uromyces pisi (Pers.) Wint., has been identified in a F2 population derived from an intraspecific cross between two wild pea (Pisum fulvum L.) accessions, IFPI3260 (resistant) and IFPI3251 (susceptible). Both parental lines and all the segregating population displayed a fully compatible interaction (high infection type), which indicates absence of hypersensitive response. Nevertheless, differences on the percentage of symptomatic area of the whole plant (disease severity) were observed. A genetic map was developed covering 1283.3 cM and including 146 markers (144 random amplified polymorphic DNA (RAPDs) and two sequence tagged sites (STSs) markers) distributed in 9 linkage groups. A QTL explaining 63% of the total phenotypic variation was located in linkage group 3. RAPDs markers (OPY111316 and OPV171078) flanking this QTL should allow, after their conversion in SCARs, a reliable marker-assisted selection for rust resistance.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pea rust has become an important pathogen of dry pea from the mid-1980s and is distributed in Europe, North and South America, Asia, Australia and New Zealand. The pathogen develops in warm, humid weather and the disease usually appears during mid-spring when the crop is at flowering or podding stage (EPPO 2009). The activity of the rust generally results in the break of physiological and biochemical processes in the plant, and photosynthesis is particularly reduced (EPPO 2009). In years of epidemics, rust strongly affects leaves dry up. Most of them fall down and pods remain undeveloped, which consequently yield losses higher than 30%.
In tropical and subtropical regions as in India and China, pea rust has been reported to be caused by the fungus Uromyces viciae-fabae (Pers.) J. Schröt (syn. U. fabae (Pers.) de Bary), since the warm humid weather is suitable for the appearance of both the uredial and the aecidial stage (Pal et al. 1980; Singh et al. 2004; Kushwaha et al. 2006). However, in temperate regions, pea seedlings can be infected by U. viciae-fabae, but the pathogen hardly settle down and progress in field conditions, being U. pisi (Pers.) Wint. the main responsible for the disease (Emeran et al. 2005; Barilli et al. 2009a, c).
U. pisi is a heteroecious macrocyclic fungus that completes its life cycle on the spontaneous Euphorbia cyparissias L. (cypress spurge) (Pilet 1952), which becomes severely infected (Pfunder and Roy 2000). The fungus overwinters on vegetation residues as teliospores and on rhizomes of Euphorbia as mycelium.
Only recently, sources of incomplete resistance to U. pisi from a Pisum spp. collection have been identified in field conditions and further characterized macro and microscopically in controlled environment (Barilli et al. 2009b, c). Higher levels of resistance have been identified in the wild species Pisum fulvum L. (Barilli et al. 2009a), but such resistance has not yet been efficiently used in breeding programs. The wild P. fulvum accession IFPI3260 has shown to have very good levels of resistance in both, field and controlled conditions (Barilli et al. 2009a, b), consisting of a pre penetration resistance not associated with host cell death.
Knowledge on the genetic system controlling resistance to U. pisi in accession IFPI3260 would facilitate gene transfer to pea cultivars. To date, the only study regarding the genetic nature of the resistance in peas against rust have been carried out on P. sativum accessions using U. viciae-fabae as pathogen (Katiyar and Ram 1987; Vijayalakshmi et al. 2005). On this pathosystem, the dominant nature of the partial resistance, assayed as number of pustules cm−2, has been justified as the expression of a single major gene for which the symbol Ruf was proposed (Katiyar and Ram 1987; Vijayalakshmi et al. 2005). Further, this trait seems to be affected by some polygenes in addition to the proposed oligogene Ruf (Singh and Ram 2001). Two random amplified polymorphic DNA (RAPD) markers, SC10-82 360 and SCRI-71 1000 were detected flanking the gene Ruf 10.8 and 24.5 cM apart, respectively, but these markers were not close enough to allow a reliable marker-assisted selection (MAS) approach for rust resistance improvement (Vijayalakshmi et al. 2005).
The use of molecular markers could accelerate plant breeding in relation to certain agronomic traits, including disease resistance, enabling combinations of resistance genes to be assembled. However, no published reports on molecular mapping of U. pisi resistance genes are available in Pisum spp. so far. Therefore, the goals of the preset work have been: (1) to develop a genetic map of P. fulvum using both RAPD and sequence tagged sites (STS) markers; (2) to detect quantitative trait loci (QTLs) for U. pisi resistance with the aim to study the genetic inheritance of the resistance to this pathogen; (3) to identify molecular markers tightly linked to the responsible gene(s) which might be useful for marker assisted selection (MAS) purposes.
Materials and methods
Plant material
The cross IFPI3260 (resistant) × IFPI3251 (susceptible) was selected for the study because the parental lines have shown a consistent reaction to rust for more than four years. Furthermore, both parents were also resistant to powdery mildew (Erysiphe pisi Syd.) and broomrape (Orobanche crenata Forsk.) (Rubiales et al. 2005; Fondevilla et al. 2007), two other important pathogens under Mediterranean conditions (Rubiales et al. 2009). This fact might facilitate future crop improvement by producing new pea cultivars with multiple resistance specificities using molecular breeding strategies. Ninety-four F2 plants were sown in the field at the beginning of February together with the susceptible control cv. “Messire”. In June, when pods were at maturity, 25 seeds per plants were collected and sown under controlled condition to obtain the subsequent F3 families.
These 94 F3 families were grown in pots (35 × 35 cm), two plants per pot which were filled with a 1:1 mixture of sand and peat. Three replicas were performed with eight plants per replica in a completely randomized design. Seedlings were inoculated when the third leaf was completely expanded. Inoculation was carried out by dusting the plants with U. pisi isolate Up-Co01 urediospores (2 mg spores plant −1) diluted in pure talc (1:10, v:v) using a spore settling tower. Plants were incubated for 24 h at 20°C in complete darkness and 100% relative humidity, then transferred to a growth chamber at 20°C, with a photoperiod of 14/10 h day/night regime, with 148 μmol m−2 s−1 irradiance at plant canopy.
Infection Type (IT) and Disease Severity (DS) were observed 15 days after inoculation. DS was visually estimated as the percentage of symptomatic area of the whole plant. IT was assessed using the 0-4 scale of Stackman et al. (1962), where IT 0 = no symptoms, IT; = necrotic flecks, IT 1 = minute pustules barely sporulating; IT 2 = necrotic halo surrounding small pustules, IT 3 = chlorotic halo and IT 4 = well-formed pustules with no associated chlorosis and necrosis.
PCR analysis
For DNA extraction, about 100 mg of young leaf tissue from the F2 plants was excised, immediately frozen in liquid nitrogen and stored at −80°C. DNA was isolated using the CTAB method (Lassner et al. 1989), with slight modifications (Torres et al. 1993). In the initial screening, a total of 660 RAPD decamer primers (prefix “OP-” from Operon Technologies, Alameda, CA, USA) were tested on the parents for polymorphism. Once a polymorphism was found, the corresponding primer was genotyped in the F2 population and the two parents for linkage analysis. Optimal reaction conditions were established according to Cobos et al. (2005). Amplification was performed in a Gene Amp PCR System 9700 (Applied Biosystems, Foster City, CA, USA) in 25-μl volumes including 20-40 ng plant genomic DNA, buffer (50 mM KCl, 10 mM Tris–HCl, 0.1% Triton X-100), 2 mM MgCl2, 2.5 mM of each dNTP, 0.2 μM of each primer and 1 unit of Taq DNA polymerase (Biotools B&M Labs, Madrid, Spain). Forty cycles denaturation at 94°C for 20 s, annealing for 1 min at 36°C and 1 min elongation at 72°C, with a final extension at 72°C for 8 min were performed.
In addition 10 STS markers reported by Gilpin et al. (1997) were surveyed for polymorphism using their protocol. The 25-μl reaction volumes included 20 ng plant genomic DNA in buffer (10 mM Tris–HCl, 50 mM KCl, 0.1% Triton X-100), 2.5 mM Mg Cl2, 200 μM each dNTP, 1.0 μM primer and 0.625 U Taq DNA polymerase (Bioline Ltd, London, UK). After DNA denaturation for 1 min at 95°C, the reaction mixture was subjected to 40 cycles of the following temperature profile: 95°C for 1 min, 60°C for 1 min and 72°C for 1 min, followed by a final extension at 72°C for 8 min. When no polymorphism was detected, PCR products from both parents were digested with a range of restriction endonucleases which recognized 4- and 5- base sequences (BioLabsinc, Ipswich, MA, USA). A 0.2 μl aliquot of restriction enzyme, 12 μl of sterile water and 2.5 μl of the buffer required for each enzyme were added to 10 μl of the PCR reaction, and the digestion was incubated overnight at 37°C.
Amplification products from the RAPD and STSs were separated in gels composed of a mixture of 1% agarose and 1% Nu-Sieve agarose (Hispanlab, Spain) in one-time Tris–borate-EDTA (TBE) buffer containing 0.5 μg/ml ethidium bromide (Sambrook et al. 1989) for 3 h at 105 V. The gel was scanned using Kodak Digital Science 1D (ver. 2.0 and 3.5) Image Analysis Software (New Haven, CT, USA).
Statistical analysis
Segregation data from the F2 population together with the parental accessions IFPI3260 and IFPI3251 were used to obtain a map. Goodness-of-fit to the expected 3:1 and 1:2:1 segregation ratio of the RAPD and STS markers, respectively, was calculated by χ2 tests. Linkage analysis was performed using MapMaker ver. 2.0 (Lander et al. 1987). A LOD score threshold of 3 and a maximum recombination fraction of 0.3 were employed as general linkage criteria to establish linkage groups (LGs). Kosambi’s function was applied to estimate map unit distances (Kosambi 1944).
The phenotypic DS values measured in controlled conditions were normalized by arcsine transformation [y = arcsine (√DS)], and used for QTL analysis. Skewness and Kurtosis coefficients were calculated following Lynch and Walsh procedure (1997). QTL analysis was performed using QTL Cartographer, ver. 2.5 (Wang et al. 2005). The threshold for the detection of a QTL appropriate for a sparse-map case (Lander and Botstein 1989) was calculated by a Bonferroni correction to ensure the experiment-wise significance level of 95%. If n independent tests (number of marker intervals) with significance level α are conducted, the probability that at least one test being false positive is (Lynch and Walsh 1997):
Bonferroni’s correction for multiple comparisons states that an overall significance level γ requires that each individual test be based on a significance level of:
In our case, 137 marker intervals were tested and the experiment-wise type-I error probability was set to α = 0.05. Thus, comparison-wise type-I error probability using Bonferroni correction was calculated (g = 0.000374) corresponding to an LR threshold of 18.34 (LOD = 3.98) with 3 degrees of freedom (additive and dominance effects for a QTL are fitted).
The coefficient of determination (R2) for the marker most tightly linked to a QTL was used to estimate the proportion of the total phenotypic variation explained by the QTL.
Results
Disease assessment
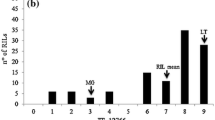
Confirming results in previous studies (Barilli et al. 2009a, b), the parental accessions showed a compatible interaction (high IT) against U. pisi although they differed in DS. IFPI3260 displayed an incomplete resistance with a DS value of 1% (arcsine transformed DS value = 0.10). In contrast, the parental IFPI3251 was highly susceptible, showing a DS value of 19.4% (arcsine transformed DS value = 0.46) (Fig. 1). Disease assessment in the F3 population followed a normal distribution (Lynch and Walsh normality test, P > 0.01), with DS values ranking between 0.4 and 14.9% (arcsine transformed DS values between 0.06 and 0.4). Highly significant differences between families (ANOVA P < 0.0001) were observed. The coefficient of Skewness in controlled conditions was of 0.86, indicating that the population distribution tends to the resistance as did the parent IFPI3260. In addition, five lines more resistant than IFPI3260 were observed, but differences were not significant (Dunnet test, P > 0.05).

Frequency distribution for rust resistance estimated as the arcsin transformed DS values in the pea F2 population IFPI3260 × IFPI3251 under controlled conditions
Linkage map
Eighteen RAPD primers out of the 660 tested in the parental lines, revealed the existence of polymorphism and were screened in the whole F2 population originating 168 clear and scorable bands. Segregation deviation from the expected 3:1 ratio was determined based on a chi-squared test at P < 0.05 and P < 0.01 levels. At P < 0.05 level, only two RAPD markers (OPAB17519 and OPAD111277) showed distorted segregation.
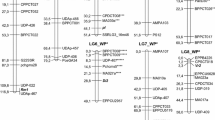
Only two of the 10 STSs analyzed showed a direct length polymorphism between the parental lines while the remaining 8 had to be digested with different restriction endonucleases to reveal six putative codominant STSs (P202, Q500 and P393 digested with HindI; M27 and Q363 with RSAI; P628 digested with AluI). However, the amplification patterns achieved in most cases were not precise and repeatable enough to genotype unambiguously the whole population. As a result, only 2 STSs (P482 and Q363) could be properly amplified in the F2 population and included in the linkage analysis. The map covered 1283.3 cM of the wild pea genome with an average inter-marker distance of 9.73 cM (Table 1). The use of two common markers (P482 and Q363) with the international pea consensus genetic map (Weeden et al. 1998) enabled two of the linkage groups (LGs 5 and 6) to be assigned to the pea chromosome VI (Fig. 2).

Genetic linkage map from 94 F2 plants from the cross IFPI3260 × IFPI3251. The size of each group is indicated in centimorgans (cM). Bar position indicate the location of the QTL for resistance to U. pisi in controlled conditions. Outer and inner interval corresponding to 1-LOD and 2-LOD support interval are indicated as a full box and a single line, respectively. Markers in bold italics are anchored to other published pea genetic maps. Markers showing distorted segregation are underlined
QTL mapping
Quantitative trait loci analysis with composite interval mapping (CIM) method revealed a single genomic region associated with resistance to U. pisi in controlled conditions located in LG 3. The putative QTL Up1 showed a LOD score of 4.83 and explained the 63% of the phenotypic variation. Up1 was localized at 32.9 cM from the beginning of the LG03, between the RAPD markers OPY111316 and OPV171078. The distance to these flanking markers was of 6 and 13.4 cM, respectively. The QTL distance to OPV171078 might be even smaller considering the 1-LOD support interval which range between 32.9 and 38.9 cM (Table 2; Fig. 3). Both markers derive from the susceptible parent IFPI3251. The resistance-enhancing allele originates from the resistant parent IFPI3260 as shown by the negative value of the additive genetic effect (-0.094); the dominant effect was of -0.056 (Table 2). Up1 showed the dominance/additive (d/a) ratio of 0.6 indicating a partially dominant gene action.

LOD profile of the QTL analysis of rust resistance obtained in the pea F2 population IFPI3260 × IFPI3251 using CIM
Discussion
In the present study, a linkage map was developed using a F2 population derived from the intraspecific cross between two Pisum fulvum lines. The approach has enabled, for the first time, the identification of a QTL for resistance to pea rust incited by U. pisi in wild pea.
The high level of resistance showed by the P. fulvum accession IFPI3260, previously reported by Barilli et al. (2009a, b), has been confirmed in the present study. This outcome points out the scoring mode for rust resistance, reported in this study, as a reliable method. Both parental lines and the derived progeny showed a compatible interaction with no associated cell death. Resistance was based on the reduction of the disease severity. This is in agreement with previous studies in which only partial resistance was described against both U. viciae-fabae (Pal et al. 1980; Singh and Sokhi 1980; Xue and Warkentin 2001; Chand et al. 2006) and U. pisi (Barilli et al. 2009a, b, c). In other legume-rust pathosystems, quantitative non-hypersensitive types of resistance are predominant (reviewed in Sillero et al. 2006), e.g. in pea—U. viciae-fabae (Pal et al. 1980), in chickpea—U. ciceris-arietini (Madrid et al. 2008) and in groundnut—Puccinia arachidis (Subrahmanyam et al. 1993). However, the hypersensitive reaction is found on certain legumes species such as Phaseolus vulgaris, against U. appendiculatus (Stavely et al. 1989), faba bean against U. viciae-fabae (Sillero et al. 2000) or soybean against Phakopsora pachyrhizi (Li 2009 ), but the great disadvantage of this type of resistance is that it is often ephemeral (Niks and Rubiales 2002), due to the evolution of virulent fungal isolates.
The map developed from the F2 population, covered a genetic distance of 1283.3 cM and contained 146 markers, distributed in nine linkage groups. The extent of genome coverage is comparable with that of previous pea maps developed in P. sativum by Gilpin et al. (1997) (1,330 cM), and higher than the maps reported by Tar’an et al. (2003) (1,274 cM), Prioul et al. (2004) and Timmermann-Vaughan et al. (2004) (1,061 and 930 cM, respectively) and Fondevilla et al. (2008) (1,214 cM). Unfortunately, our map is only connected to the pea consensus map of Weeden et al. 1998 (nearly 800 cM) by two common STS markers. The transferability to these markers allowed us to assign two linkage groups to a specific pea chromosome. In particular, the STSs P482 and Q363 located on the LG 5 and 6, respectively, enabled us to assign these LGs to the chromosome 6 of the P. sativum consensus map reported by Weeden et al. (1998).
A single QTL (Up1) was identified on LG 3. Frequency distribution for rust resistance in the pea F2 population IFPI3260 × IFPI3251 tended towards a bimodal distribution of DS values suggesting an oligogenic control of resistance in these materials. The involvement of a major genetic factor for partial resistance to rust would explain such behaviour as supported by the high R2 (0.63) revealed for the only QTL detected. The remaining variation not explained by Up1 could be due to undetected additive QTLs with weak effects or to incomplete map coverage. Analysis of a larger populations or a more precise quantitative scoring system of the disease reaction may allow the detection of additional minor QTLs that account for the remaining unexplained variance.
The bimodal distribution of the resistant trait in the progeny and the identification of a single genomic region associated with resistance to U. pisi points towards the quantitative nature of rust resistance in cross IFPI3260 × IFPI3251, and identify molecular markers associated with partial resistance. Partial resistance is usually assumed to be poligenically inherited. However, this is not always the case as single genes, causing partial resistance to rust, have been described in pea (Vijayalakshmi et al. 2005), chickpea (Madrid et al. 2008) and wheat, e.g. Lr34 (Rubiales and Niks 1995) and Lr46 (Singh et al. 1998; Martínez et al. 2001), always related to pre-haustorial resistance.
Our results indicate that the QTL explained a very high percentage of the phenotypic variation throughout the population, suggesting that effective selection could be possible with a few markers tightly linked to the QTL. The RAPD markers OPY111316 and OPV171078 are located at the position 26.9 and 46.3 cM respectively, delimiting a region fairly close to Up1. Therefore the RAPD markers OPY111316 and OPV171078 should allow, after their conversion in SCARs markers, a reliable marker-assisted selection for rust resistance.
Nevertheless, QTL mapping is only the first step in MAS, because QTL analysis needs to be refined to determine the precise position and effects of the QTLs involved. Future research will be focused on the saturation of the region bearing this QTL with more robust and transferable markers such as new STSs, microsatellites (Tauz and Renz 1984; Loridon et al. 2005) with the aim to identify one in coupling phase with the resistant parent IFPI3260, tightly linked to Up1.
Markers associated with rust resistance in P. fulvum could be also assayed in other P. sativum populations (Vijayalakshmi et al. 2005) or even in other legume species. DNA markers that allow cross-species mapping (referred as anchor markers) are critical for comparative genome analysis. The approach has revealed significant transferability between species (Gutiérrez et al. 2005; Madrid et al. 2008). This transferability could help us to verify if the same region is presented in this model specie whose genome is being sequenced.
Moreover, ESTs (Expressed Sequence Tagged Sites) derived from protein-encoding regions in a number of model species are being extensively used to detected orthologous loci in multiple legume genomes (Choi et al. 2006). Efforts to integrate the informative of anchor markers into crop legumes linkage maps are also in progress (Sato et al. 2005; Nelson et al. 2006; Phan et al. 2007; Ellwood et al. 2008). It is anticipated that the inclusion in this maps of genes of known function, will help us to verify if any of the mapped genes is a good functional or positional candidate for the rust resistance.
Our results for rust resistance must be validated across diverse locations and genetic backgrounds before the application of MAS in pea breeding programs. For this reason, we are at present developing the recombinant inbred lines (RILs) derived from the cross, as well as between U. pisi-resistant P. sativum lines and susceptible high-yielding commercial varieties, in order to analyze QTL stability in different environments and pedigrees. As susceptibility of Pisum spp. accessions to faba bean rust (U. viciae-fabae) has been already described (Pal et al. 1980; Xue and Warkentin 2001; Chand et al. 2006), the availability of RILs is also of great importance to evaluate the fixed material with different pathogen species.
In conclusion, the present study is the first report on the detection of a QTL controlling a high percentage of the resistance against U. pisi in P. fulvum and therefore may be useful to predict the location of the homologous region in related species. The inclusion of new transferable markers and the transformation into SCARs of the RAPDs tightly linked to the detected QTL will enhance the possibility of developing a map useful not only for MAS purposes in wild and cultivated peas but also for comparative mapping studies among different legumes.
References
Barilli E, Sillero JC, Fernández-Aparicio M, Rubiales D (2009a) Identification of resistance to Uromyces pisi (Pers.) Wint. in Pisum spp. germplasm. Field Crop Res 114:198–203
Barilli E, Sillero JC, Rubiales D (2009b) Characterization of resistance mechanisms to Uromyces pisi in pea. Plant Breed 128:665–670
Barilli E, Sillero JC, Serrano A, Rubiales D (2009c) Differential response of pea (Pisum sativum) to rusts incited by Uromyces viciae-fabae and U. pisi. Crop Prot 28:980–986
Chand R, Srivastava CP, Singh BD, Sarode SB (2006) Identification and characterization of slow rusting components in pea (Pisum sativum L.). Genet Resour Crop Evol 53:219–224
Choi HK, Luckow MA, Doyle J, Cook DR (2006) Development of nuclear gene-derived molecular markers linked to legume genetic maps. Mol Genet Genomics 276:56–70
Cobos MJ, Fernández MJ, Rubio J, Kharrat M, Moreno MT, Gil J, Millán T (2005) A linkage map in chickpea (Cicer arietinum L.) in two populations from Kabuli × Desi crosses: location of a resistance gene for fusarium wilt race 0. Theor Appl Genet 110:1347–1353
Ellwood SR, Phan HTT, Jordan M, Hanes J, Torres AM, Ávila CM, Cruz-Izquierdo S, Oliver RP (2008) Construction of a comparative genetic map in faba bean (Vicia faba L.) conservation of genome structure with Lens culinaris. BMC Genomics 9:380. doi:10.1186/1471-2164-9-380
Emeran AA, Sillero JC, Niks RE, Rubiales D (2005) Morphology of infection structures help to distinguish among rust fungi infecting leguminous crops. Plant Dis 89:17–22
Fondevilla S, Carver TLW, Moreno MT, Rubiales D (2007) Identification and characterization of sources of resistance to Erysiphe pisi Syd. in Pisum spp. Plant Breed 126:113–119
Fondevilla S, Satovic Z, Rubiales D, Moreno MT, Torres AM (2008) Mapping of quantitative trait loci for resistance to Mycosphaerella pinodes in Pisum sativum subsp. syriacum. Mol Breed 21:439–454
Gilpin BJ, McCallum JA, Frew TJ, Timmerman-Vaughn GM (1997) A linkage map of the pea (Pisum sativum L.) genome containing cloned sequences of known function and expressed sequence tags (ESTs). Theor Appl Genet 114:59–66
Gutiérrez MV, Vaz Patto MC, Huguet T, Cubero JI, Moreno MT, Torres AM (2005) Cross-species amplification of Medicago truncatula microsatellites across three major pulse crops. Theor Appl Genet 110:1210–1217
Katiyar RP, Ram RS (1987) Genetics of rust resistance in pea. Indian J Genet 47:46–48
Kosambi DD (1944) The estimation of map distances from recombination values. Ann Eugenics 12:461–465
Kushwaha C, Chand R, Srivastava C (2006) Role of aeciospores in outbreaks of pea (Pisum sativum) rust (Uromyces fabae). Eur J Plant Pathol 115:323–330
Lander ES, Botstein D (1989) Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121:185–199
Lander ES, Green P, Abrahamson J, Barlow A, Daly MJ, Lincoln SE, Newburg L (1987) MAPMAKER: an interactive computer program for constructing genetic linkage maps of experimental and natural populations. Genomics 1:174–181
Lassner MW, Peterson P, Yoder JI (1989) Simultaneous amplification of multiple DNA fragments by polymerase chain reaction in the analysis of transgenic plants and their progeny. Plant Mol Biol Report 7:116–128
Li S (2009) Reaction of soybean rust-resistant lines identified in Paraguay to Mississippi isolates of Phakopsora pachyrhizi. Crop Sci 49:887–894
Loridon K, Mcphee K, Morin J, Dubreuil P, Pilet-Nayel ML, Aubert G, Rameau C, Baranger A, Coyne C, Lejeune-Hènaut I, Burstin J (2005) Microsatellite marker polymorphism and mapping in pea (Pisum sativum L.). Theor Appl Genet 111:1022–1031
Lynch M, Walsh B (1997) Genetics and analysis of quantitative traits. Sinauer Associated Inc., Sunderland
Madrid E, Rubiales D, Moral A, Moreno MT, Millan T, Gil J, Rubio J (2008) Mechanism and molecular markers associated with rust resistance in a chickpea interspecific cross (Cicer arietinum × Cicer reticulatum). Eur J Plant Pathol 121:43–53
Martínez F, Niks RE, Singh RP, Rubiales D (2001) Characterization of Lr46, a gene conferring partial resistance to wheat leaf rust. Hereditas 135:111–114
Nelson M, Phan H, Ellwood S, Moolhuijzen P, Hane J, Williams A, O’Lone C, Fosu-Nyarko J, Scobie M, Cakir M, Jones M, Bellgard M, Ksiazkiewicz M, Wolko B, Barker S, Oliver R, Cowling W (2006) The first gene-based map of Lupinus angustifolius L.—location of domestication genes and conserved synteny with Medicago truncatula. Theor Appl Genet 113:225–238
Niks RE, Rubiales D (2002) Potentially durable resistance mechanisms in plant to specialized fungal pathogens. Euphytica 124:201–216
Pal AB, Brahmappa HS, Rawal RD, Ullasa BA (1980) Field resistance of pea germplasm to powdery mildew (Erysiphe polygoni) and rust (Uromyces fabae). Plant Dis 64:1085–1086
Pfunder M, Roy BA (2000) Pollinator-mediated interactions between a pathogenic fungus, Uromyces pisi (Pucciniaceae), and its host plant, Euphorbia cyparissias (Euphorbiaceae). Am J Bot 87:48–55
Phan HTT, Ellwood SR, Hane JK, Ford R, Materne M, Oliver RP (2007) Extensive macrosynteny between Medicago truncatula and Lens culinaris ssp. culinaris. Theor Appl Genet 114:549–558
Pilet PE (1952) Etude physiologique du parasitisme de l’Uromyces pisi (Pers.) de By., sur l’Euphorbia cyparissias L. Experientia 9:300–302
Prioul S, Frankevits A, Deniot G, Morin G, Baranger A (2004) Mapping of quantitative trait loci for partial resistance to Mycosphaerella pinodes in pea (Pisum sativum L.) at the seedling and adult stage. Theor Appl Genet 108:1322–1334
Rubiales D, Niks RE (1995) Characterization of Lr34, a major gene conferring nonhypersensitive resistance to wheat leaf rust. Plant Dis 79:1208–1212
Rubiales D, Moreno MT, Sillero JC (2005) Search for resistance to crenata broomrape (Orobanche crenata) in pea germplasm. Genet Res Crop Evol 52:853–861
Rubiales D, Fernández-Aparicio M, Moral A, Barilli E, Sillero JC, Fondevilla S (2009) Disease resistance in pea (Pisum sativum L.) types for autumn sowings in Mediterranean environments—a review. Czech J Genet Plant Breed 45:135–142
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual. Cold Spring Harbour Laboratory Press, New York
Sato S, Isobe S, Asamizu E, Ohmido N, Kataoka R, Nakamura Y, Kaneko T, Sakurai N, Okumura K, Klimenko I, Sasamoto S, Wada T, Watanabe A, Kohara M, Fujishiro T, Tabata S (2005) Comprehensive structural analysis of the genome of red clover (Trifolium pratense L.). DNA Res 12:301–336
Sillero JC, Moreno MT, Rubiales D (2000) Characterization of new sources of resistance to Uromyces viciae-fabae in a germplasm collection of Vicia faba. Plant Pathol 49:389–395
Sillero JC, Fondevilla S, Davidson J, Vaz Patto MC, Warketin TD, Thomas J, Rubiales D (2006) Screening techniques and sources of resistance to rusts and mildews in grain legumes. Euphytica 147:255–267
Singh R, Ram H (2001) Inheritance of days to flowering and rust resistance in peas. Res Crops 2:414–418
Singh SJ, Sokhi SS (1980) Evaluation of pea cultivars to Uromyces viciae-fabae. Plant Dis 64:671–672
Singh R, Kazi M, Huerta-Espino J (1998) Lr46, a gene conferring slow rusting resistance to leaf rust in wheat. Phytopathology 88:890–894
Singh RA, De RK, Chaudhary RG (2004) Influence of spray time of mancozeb on pea rust caused by Uromyces viciae-fabae. Indian J Agric Sci 74:502–504
Stackman EC, Stewart DM, Loegering WQ (1962) Identification of physiologic races of Puccinia graminis var. tritici. USDA, Agricultural Research Service. E617, Washington
Stavely JR, Steadman JR, McMillan RT (1989) New pathogenic variability in Uromyces appendiculatus in North America. Plant Dis 73:428–432
Subrahmanyam P, McDonald D, Reddy LJ, Nigam SN, Smith DH (1993) Origin and utilization of rust resistance in groundnut. In: Jacobs T, Parlevliet JE (eds) Durability of disease resistance. Kluver Academic Publishers, Dordrecht, pp 147–158
Tar’an B, Warkentin T, Somers DJ, Miranda D, Vandenberg A, Balde S, Woods S, Bing D, Xue A, DeKoeyer D, Penner G (2003) Quantitative trait loci for lodging resistance. Plant height and partial resistance to mycosphaerella blight in field pea (Pisum sativum L.). Theor Appl Genet 107:1482–1491
Tauz D, Renz M (1984) Simple sequences are ubiquitous repetitive components of eukaryotic genomes. Nucleic Acids Res 12:4127–4138
Timmermann-Vaughan GM, Frew TJ, Butler R, Murray S, Gilpin M, Falloon K, Johnston P, Lakeman MB, Russell AC, Khan T (2004) Validation of quantitative trait loci for Ascochyta blight resistance in pea (Pisum sativum L.), using populations from two crosses. Theor Appl Genet 109:1620–1631
Torres AM, Weeden NF, Martín A (1993) Linkage among isozyme, RFLP and RAPD markers in Vicia faba. Theor Appl Genet 85:937–945
Vijayalakshmi S, Yadav K, Kushwaha C, Sarode SB, Srivastava CP, Chand R, Singh BD (2005) Identification of RAPD markers linked to the rust (Uromyces fabae) resistance gene in pea (Pisum sativum). Euphytica 144:265–274
EPPO Standards Pea http://archives.eppo.org/EPPOStandards/PP2_GPP/pp2-14-e.doc. Visited 13 November 2009
Wang S, Basten CJ, Gaffney P, Zeng ZB (2005) Windows QTL cartographer version 2.5. Statistical genetics. North Carolina State University, Raleigh
Weeden NF, Ellis THN, Timmerman-Vaughan GM, Swiecicki WK, Rozov SM, Berdnikov VA (1998) A consensus likage map for Pisum sativum. Pisum Genetics 30:1–4
Xue AG, Warkentin TD (2001) Reaction of field pea varieties to three isolates of Uromyces fabae. Plant Sci 82:253–255
Acknowledgments
We thank N. Gutiérrez, C. Avila, L. Madrid and G. Lara Santos for good advice and technical support in the laboratory. Authors are greatly indebted to Spanish AGL2008-01239 and European Union FP6-2002-FOOD-1-506223 projects for financial support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Barilli, E., Satovic, Z., Rubiales, D. et al. Mapping of quantitative trait loci controlling partial resistance against rust incited by Uromyces pisi (Pers.) Wint. in a Pisum fulvum L. intraspecific cross. Euphytica 175, 151–159 (2010). https://doi.org/10.1007/s10681-010-0141-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10681-010-0141-z