
Abstract
To prevent lodging and sprouting damage in rye, a dominant dwarfing gene, Ddw1, has been incorporated into breeding lines to reduce plant height in Finland. However, the inability to identify heterozygous plants makes it difficult to purify breeding lines. Doubled haploidy (EM-1 × Voima) and bulked segregant analysis were used to search for an efficient PCR-based tool to identify homozygous short plants. In addition, SNP (single nucleotide polymorphism) markers were created from the endosperm-specific β-amylase gene and the microsatellite locus REMS1218 expressed in rye and known to be located near Ddw1. The best marker was a combination of the microsatellite REMS1218 and the SNP created from it, located 13 cM from the QTL corresponding to Ddw1. However, for a rye breeder, the SNP alone is an adequate tool to identify plants homozygous for the EM-1 allele and it can be used in selection for the desirable growth habit in rye.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rye (Secale cereale L., 2n = 2x = 14), an out-crossing species, is mainly cultivated in the northern and eastern part of Europe and is the second most important bread cereal after wheat in Finland. Typically in Finland, winter rye cultivars are tolerant to freezing and resistant to low-temperature fungal infections. However, these cultivars with long straw are lodging-sensitive and susceptible to pre-harvest sprouting, which decreases the quality of rye bread flour.
Height reduction in cereals is achieved by dwarfing genes, which are divided into two groups: gibberellin (GA) insensitive (unresponsive to exogenously applied GA) and sensitive (Börner et al. 1996). In main cereals, many dwarfing genes (e.g., wheat: Rht-B1b, Rht-D1b, Rht8, Rht9; barley: denso or sdw1, Gpert; oat: Dw6, Dw7) have been incorporated into European or American germplasm and used in cultivar development (Marshall and Murphy 1981; Milach et al. 1998; Milach and Federizzi 2001). It has been observed that dwarfing genes may have pleiotropic effects on other traits. For example, Rht3 gene reduces the synthesis of α-amylase during germination and therefore prevents pre-harvest sprouting damage in wheat (Milach and Federizzi 2001).
Two dominant GA sensitive dwarfing genes, Ddw1 and Ddw2, and nine recessive, sensitive or insensitive, genes have been discovered in rye (Börner et al. 1996). Ddw1 was discovered in the natural dwarf mutant EM-1 of the Vavilov genebank collection in St. Petersburg (Börner et al. 1996; Kobyliansky 1972), and it is homoeologous to the dwarfing gene Rht12 in wheat (Börner et al. 1996, 1998; Korzun et al. 1997). Ddw1 was originally named as Humilus (Hl, Sturm and Engel 1980), and it is located on the long arm of 5R, between the genes for hairy peduncle (Hp1) and β-amylase (Korzun et al. 1996). The two closest markers found near Ddw1 are the wheat RFLP marker Xwg199 and the isozyme marker β-amy-R1 (Börner et al. 1999; Korzun et al. 1996). As the best-known short straw mutant, EM-1 has been included in many Eastern European, as well as Finnish, rye breeding programs to prevent lodging in rye (Milach and Federizzi 2001). However, purifying lines from heterozygous plants has been impossible due to the indistinguishable phenotypes of homozygous and heterozygous plants having a short growth habit. In order to trace favourable alleles, marker assisted selection (MAS) is often used in modern breeding (Francia et al. 2005). The aim of this study was to develop an efficient PCR-based DNA marker linked to the dwarfing gene Ddw1. Doubled haploidy, bulked segregant (BSA, Michelmore et al. 1991), and SNP (single nucleotide polymorphism) analyses were used for tagging the gene.
Materials and methods
Plant material and height measurement
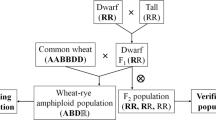
Doubled haploid (DH) plants were developed from the Finnish tall winter rye cv. Voima and the Russian short mutant EM-1 with the dominant dwarfing gene Ddw1 (Kobyliansky 1972) using anther culture (Tenhola-Roininen et al. 2006). The plants were grown as previously described (Tenhola-Roininen et al. 2005). Two populations (109/49 individuals) segregating for short straw were created (Fig. 1; Tenhola-Roininen et al. 2006). Only the parents of the populations were DHs because the yield of F1 seeds was too low for use of anther culture to create a mapping population. Two different genotypes of the DH parents were used to lessen the effects of inbreeding depression in F1 crosses. The larger population was used to find markers linked to Ddw1 (hereafter referred to as the mapping population). Promising DNA markers were then analysed also in the smaller population, and the results from the two populations were pooled. The height of the plants just before ripening was measured in the greenhouse from the soil surface to the top of the tiller (with the spike). The height of an individual was calculated as the mean of the three highest tillers.

Production of rye populations segregating for short straw: a crossing scheme
DNA isolation and bulking of individuals
DNAs were extracted from 2 g of young leaves by a modified CTAB (cetyltrimethyl ammonium bromide) II method (Poulsen et al. 1993). The crushed leaves were incubated in the CTAB isolation buffer at 60°C for max 60 min. The chloroform: isoamyl alcohol suspension was added and mixed with the extracted leaves before centrifugation at +6°C in the cold room. 10 μg/ml RNAse (Sigma–Aldrich, St. Louis, MO, USA) was added to the aqueous phase. The samples were incubated at +37°C for 30–60 min and on ice for 5 min. After cold isopropanol precipitation, nucleic acids were transferred to new Eppendorf tubes, washed twice with cold 70% ethanol and centrifuged at 10,000 g for 10 min. Air-dried DNA pellets were re-dissolved in TE buffer (10 mM Tris–HCl, 1 mM EDTA, pH 8.0). BSA was used to find polymorphic markers between ‘short’ and ‘tall’ bulks which consisted of the DNA samples of the nine shortest and the nine tallest individuals of the mapping population.
DNA marker analyses
RAPD (random amplified polymorphic DNA) primers were either synthesized on an Applied Biosystems 392 DNA/RNA Synthesizer or purchased from Operon Technologies (Alameda, California, USA). For one RAPD primer, APR5.2 (5′-AGAGATCTCC-3′), which was previously found to be located on 5RL, the PCR program of Masojć et al. (2001) was used. Other RAPD analyses were carried out as follows: A reaction volume of 25 μl contained 10–30 ng DNA, 10–15 pmol each primer, 100 μM each dNTP, 1.5 mM MgCl2, 0.75–1 U Taq DNA polymerase (MBI Fermentas, Vilnius, Lithuania, or Red Hot®, ABgene, Surrey, UK), and buffer supplied by the enzyme manufacturer. The amplifications were carried out with the profile of 95°C for 3 min 30 s, followed by 40 cycles of 95°C for 30 s, 35°C for 30 s and 72°C for 2 min, and with a final extension at 72°C for 4 min.
Various exon-intron-based primers or primer combinations (ET and IT primers: sequence data provided from Andrzej Rafalski, IHAR, Poland; SRAP, sequence related amplified polymorphism: Li and Quiros 2001) were amplified in a PCR reaction volume of 20 μl or 25 μl containing 20–40 ng DNA, 10–20 pmol primers, 100 μM each dNTP, 1.5–2 mM MgCl2, 0.4–0.5 U Taq DNA polymerase (Red Hot®, ABgene, Surrey, UK, or Biotools B&M Labs, Madrid, Spain), and the buffer supplied by the enzyme manufacturer. The PCR program with ET and IT primers was carried out according to Rafalski et al. (2002) with the addition of a denaturation step at 95°C for 3 min 30 s and a final extension step at 72°C for 10 min. The SRAP markers were amplified using an annealing temperature of 47°C for 40 cycles but otherwise as described by Li and Quiros (2001).
Seventy-one rye microsatellites (SCM) developed in BAZ (Bundesanstalt für Züchtungsforschung an Kulturpflanzen, Germany; Hackauf and Wehling 2002b), 6 rye microsatellites (RMS) from Lochow-Petkus GmbH and Hybro GmbH & CoKG, Germany, 11 rye SCMs, and one wheat microsatellite, WMS6, described by Saal and Wricke (1999) and Röder et al. (1995), repectively, 47 microsatellites from barley and wheat (Becker and Heun 1995; Liu et al. 1996; Ramsay et al. 2000; Röder et al. 1998), and one rye expressed microsatellite site, REMS1218 (GenBank accession no. BE587316), known to be located on 5RL (Khlestkina et al. 2004), were used in the study. All the primers, except those provided from BAZ, were either synthesized on an Applied Biosystems 392 DNA/RNA Synthesizer or purchased from Sigma Genosys, UK. The SCM primer pairs from BAZ were amplified with a three-primer system (a forward, a reverse, and a M13 primer; Oetting et al. 1995) of which the M13 primer was labelled with a fluorescent dye (Cy-5, FAM, FITCH, HEX or TET) and the forward primer had a M13 tail. The other microsatellites were amplified with a two-primer system, the forward primer carrying a fluorescent dye. The PCR reactions were carried out as follows: A reaction volume of 12.5, 20, or 25 μl, contained 20–40 ng DNA, 2.5–10 pmol forward primer, 5–10 pmol reverse primer, 1.0–1.5 pmol labelled M13 primer if used, 50 or 100 μM each dNTP, 1.5–2 mM MgCl2, 0.2–0.5 U Taq DNA polymerase (Red Hot®, ABgene, Surrey, UK, or HotStarTaq plus Q-solution, Qiagen, Valencia, CA, USA, or Biotools B&M Labs, Madrid, Spain), and the buffer supplied by the enzyme manufacturer.
The different microsatellites were amplified with various PCR programs to get optimal results. For the SCMs from BAZ, five different PCR programs were used. We started with a program described by Khlestkina et al. (2004) with the modification of an initial denaturation step of 3 min 30 s and the cycles increased from 27 to 35 at 47°C for 1 min 15 s. The other programs were modifications of the preceding PCR program. The RMS microsatellites were amplified as follows: a denaturation step at 94°C for 2 min, followed by 30–45 cycles of 94°C for 15 s, annealing (52, 60 or 64°C) for 30 s, 72°C for 2 min, and a final extension at 72°C for 5 min. The REMS1218 (Khlestkina et al. 2004) primers (forward, 5′-CGC ACA AAC AAA AAC ACG AC-3′; reverse, 5′-CAA ACA AAC CCA TTG ACA CG-3′) were designed using a PRIMER 3.0 program (http://frodo.wi.mit.edu, Rozen and Skaletsky 2000). The PCR program was as follows: 95°C for 2 min, followed by 38 cycles of 95°C for 30 s, 55°C for 30 s, 72°C for 1 min, and a final extension step at 72°C for 6 min. Other rye microsatellites, as well as barley and wheat microsatellites were amplified according to published PCR programs (Becker and Heun 1995; Liu et al. 1996; Ramsay et al. 2000; Saal and Wricke 1999) with minor modifications.
All PCR amplifications were run on a Mastercycler® (Eppendorf, Hamburg, Germany) or a PTC-220 DNA Engine DYAD™ Peltier Thermal Cycler (MJ Research, Watertown, Mass., USA). Microsatellites and SRAPs were detected with an A.L.F. DNA Sequencer™ (Ge Healthcare, Buckinghamshire, UK), an ALFexpress™ DNA Sequencer (Ge Healthcare, Buckinghamshire, UK), or a MegaBACE™ 500 Sequencer (GE Healthcare, Buckinghamshire, UK), and the data was analysed using a Fragment Analyser 1.02 (Amersham Pharmacia Biotech) or a MegaBACE™ Genetic Profiler 2.2 (Amersham Biosciences).
SNP analysis for REMS1218
For sequencing, the microsatellite site REMS1218 was amplified from two DH parents of the mapping population with the same forward primer as for the REMS1218 microsatellite, but with the reverse primer 5′-TTG GGG TTG AAT TGG AAG TG-3′ to increase the size of the sequenced fragment. The segment was amplified as follows: 95°C for 2 min, followed by 38 cycles of 95°C for 30 s, 62°C for 30 s, 72°C for 1 min, and a final extension step at 72°C for 6 min. The amplified DNA fragments were purified using a GFX™ PCR DNA and Gel Band Purification Kit (Amersham Biosciences, Uppsala, Sweden) or NucleoSpin® Extract II Kit (Macherey-Nagel GmbH & Co. KG, Düren, Germany). Fragments were ligated into the pCR®-TOPO® vector and transformed into E. coli TOP10 cells with the TOPO TA cloning Kit for Sequencing (Invitrogen, Carlsbad, CA, USA). Plasmid DNAs were purified with the CONCERT Rapid Plasmid Miniprep System (Life Technologies, Carlsbad, CA, USA) or the Fast Plasmid Mini Kit (Eppendorf, Hamburg, Germany). Several clones from both parents were sequenced using a MegaBACE™ 500 Sequencer (GE Healthcare, Buckinghamshire, UK).
Using the sequence information, a SNP primer (5′-CTT ACA TGT CAC ATC TCC T-3′), which identifies a nucleotide difference A/C between EM-1 and Voima: in an intron, was designed. In the SNP analysis, the fragments were amplified with the same primers as for the cloning. The PCR reaction was carried out as described for the microsatellites, except that the reaction contained 100 ng DNA and 5 mM MgCl2. The PCR amplification was performed as follows: 2 min at 95°C, followed by 45 cycles of 95°C for 30 s, 60°C for 30 s, 72°C for 1 min 30 s, and a final extension step at 72°C for 5 min. The SNP reaction was carried out using the MegaBACE SnuPe Genotyping Kit (Amersham Biosciences, Uppsala, Sweden). The reaction products were run using a MegaBACE™ 500 Sequencer (GE Healthcare, Buckinghamshire, UK) and analysed with the MegaBACE™ SNP Profiler, version 1.0 (GE Healthcare, Buckinghamshire, UK).
SNP analysis for β-amylase
Because the rye endosperm-specific beta-amylase gene, β-amy1, is located near Ddw1 on 5RL (Korzun et al. 1996), PCR primers were developed according to the sequence of β-amy1 (D499999) in barley to amplify the corresponding gene from rye and to develop a SNP marker. For direct sequencing, the gene was amplified from intron 3 to intron 6 with two primer pairs (5′-AT GGT CGT TCC CAG GCA TCG and 5′-GGC TAA GGG AAC TCG CAT TG-3′; 5′-GGC AAT CAA GGT ATA AGC AC-3′ and 5′-ACA TAT GGG TCA CGA GGC TG-3′) from the DH parents. The PCR reaction volume of 20 μl contained 20 ng DNA, 5 pmol primers, 50–100 μM each dNTP, 1.5 mM MgCl2, 0.2–1 U Taq DNA polymerase (HotMaster™, Eppendorf, Hamburg, Germany or MBI Fermentas, Vilnius, Lithuania, or Red Hot®, ABgene, Surrey, UK) and the buffer supplied by the enzyme manufacturer. The PCR program was as follows: 94°C or 95°C for 2 min, followed by 40 cycles of 94°C for 1 min, 53°C or 58°C for 30 s, 65°C or 72°C for 2 min, and a final extension step of 65°C or 72°C for 10 min. The amplified DNA fragments were purified using a GFX™ PCR DNA and Gel Band Purification Kit (Amersham Biosciences, Uppsala, Sweden). Both parents were sequenced using a MegaBACE™ 500 Sequencer (GE Healthcare, Buckinghamshire, UK).
For SNP analysis, the forward primer was designed so that the last base was different from the one in the rye ubiquitous β-amylase (the enzyme is located in vegetative organs) gene. This was to prevent amplifying the wrong gene, given the high similarity between the endosperm-specific and the ubiquitous β-amylase (Sadowski et al. 1993). The PCR reaction used the primers 5′-GTG CTA TGA TAA GTA CCT GC-3′ (forward) and 5′-GGC TAA GGG AAC TCG CAT TG-3′ (reverse) as otherwise described above. The PCR program was as follows: 95°C for 2 min, followed by 35 cycles of 95°C for 30 s, 53°C for 30 s, 72°C for 1 min, and a final extension step at 72°C for 6 min. Based on a difference (A/G) in the sequences of the parents (EM-1 and Voima, respectively) in exon 4, the SNP reaction was run with the SNP primer (5′-GTC ACC GTG CTT GAT CAG-3′) using the MegaBACE SnuPe Genotyping Kit (Amersham Biosciences, Uppsala, Sweden). The PCR products were run using a MegaBACE™ 500 Sequencer (GE Healthcare, Buckinghamshire, UK) and analysed with the MegaBACE™ SNP Profiler, version 1.0 (GE Healthcare, Buckinghamshire, UK).
Statistical, mapping and comparative analyses
The association of markers with plant height was analysed using the ANOVA or GLM statistical procedures (SAS Institute inc., version 9.1, Cary, NC, USA 2004) with a significance level of 5% (SAS Institute inc., version 9.1, Cary, NC, USA 2004). The MAPMAKER 3.0 software (Lander et al. 1987) with a LOD (logarithm of odds) value of 3.0 and a maximum recombination fraction criterium of 0.4 was used for determining linkages between markers. Genetic distances in centiMorgans (cM) were calculated by a Haldane’s mapping function (Haldane and Smith 1947). The location of Ddw1 was determined using a MAPMAKER/QTL 1.1 program. BLASTX/BLASTN searches (Altschul et al. 1997) against GenBank databases were performed using the National Center for Biotechnology Information server (Bethesda, Md., USA; http://www.ncbi.nlm.nih.gov).
Results
Distribution of plant height
The heights of the DH parents of the two populations (a crossing scheme in Fig. 1) were 66.5 and 79.4 cm for EM-1 (genotypes 1 and 2, respectively), and 95.3, 93.5, 100.0, and 95.9 cm for Voima (3 clones from genotype 1 and one clone from genotype 2, respectively). In the mapping population, the mean heights of the two F1 populations were 78.2 and 104.8 cm, but were 74 and 75.9 cm in the smaller population. DH plants are generally shorter than normal heterozygous plants (Tenhola-Roininen et al. 2006) thus no comparison between the heights of DHs and F1 populations can be done. The plant heights in the mapping population varied from 69 to 179 cm (mean 117.4 ± 29.4 cm, Fig. 2) and in the smaller population from 74.2 to 155 cm (mean 109.5 ± 21.1 cm). The distribution of heights in the populations seems to indicate a monogenic dominant inheritance of the dwarfing gene, Ddw1. Besides Ddw1, also other minor quantitative loci as well as environment cause variation in heights in these populations. In the tall and short bulks of the mapping population, the mean heights of the nine individuals were 171.1 and 75.9 cm, respectively.

Distribution of different genotypes of the combined marker, REMS1218com, in different height classes of the rye mapping population (black EM-1 homozygote; light Voima homozygote; grey heterozygote). Some individuals classified as heterozygotes might actually be homozygous for EM-1
Polymorphic markers and their association with plant height
BSA analysis was used to find polymorphic markers linked to short straw (Table 1). The primers or primer combinations were tested either first in the bulks and then in the parents (with RAPD markers) or vice versa (with microsatellite markers), or at the same time both in the bulks and in the parents (with exon-intron-based primers). In total, 589 markers were tested, following which 14 polymorphic markers were applied to the whole mapping population.
Less than half of the RAPD markers were polymorphic in the bulks (Table 1). Sixty-two robust primers that were polymorphic also in the parents were screened in the separate individuals of the bulks. Five markers appeared to be associated with height and were tested in the mapping population. One RAPD marker (OPR-06780) was clearly associated with straw height (Table 2). Three of the analysed exon-intron-based primers were polymorphic in the parents and in the bulks and were analysed in each individual of the bulks. One of them (ET2280) seemed to be associated with height and indeed showed a significant association (Table 2) when tested in the mapping population.
Both rye microsatellites known to be located on chromosome 5R and those, which had not been localized but may nevertheless be on 5R were used in the study. About 40% of the rye microsatellites tested were polymorphic in the parents (Table 1), and of them the most robust ones were tested in the bulks. The separate individuals of the bulks were tested with 16 rye microsatellites, and four of them were analysed in the mapping population.
In addition, microsatellite REMS1218, known to be located near Ddw1, was analysed in the whole population. The primers amplified two fragments (317 and 321 bp), of which only the longer one (derived from EM-1) was polymorphic. Therefore, the microsatellite was analysed as a dominant marker and it showed the strongest association with plant height among all the analysed markers (Table 2). When the visible marker allele is associated with short straw, a dominant marker cannot recognise the homozygotes for the EM-1 allele, which are necessary for breeding purposes. Therefore, we wanted to convert REMS1218 into a codominant SNP marker. The microsatellite REMS1218 site was amplified and sequenced from one of the EM-1 and one of the Voima DH parents, and several clones were analysed. In Voima, all the clones have the same number of GA repeats (the size of the amplified fragment 317 bp) whereas in EM-1, two types of clones were discovered: one with 5 repeats (fragment size 317 bp) and the other with 7 repeats (321 bp). In addition to differences in repeat number, there were a few nucleotide differences between the two EM-1 clone types as well as between Voima clones. One difference between the EM-1 and Voima genotypes was found in an intron region (128 bp). Based on this difference, a SNP marker was generated. However, the SNP marker can differentiate between the EM-1 and Voima genotypes but not between the two different loci amplified by the REMS1218 microsatellite primers. Nevertheless, by combining the results (REMS1218com) of SNP and microsatellite analyses, the genotypes could be scored correctly. The scoring scheme is outlined in Fig. 3: (a) when the SNP analysis identified the Voima allele and the microsatellite analysis showed only a 317 bp fragment, the plant was a Voima homozygote; (b) when the SNP analysis showed the EM-1 allele and the microsatellite primers amplified two fragments, the individual was an EM-1 homozygote; (c) when the SNP analysis displayed both alleles, but the microsatellite analysis only a 317 bp fragment, the individual was a Voima homozygote because then the EM-1 allele originated from the other locus; (d) when the SNP analysis displayed both alleles, and also two microsatellite fragments were amplified, the interpretation was ambiguous because the plant could be either an EM-1 homozygote (only a minor fraction of the group) or a heterozygote depending on where the Voima allele was derived from, the correct or the wrong locus.

The combined results (REMS1218com) from the SNP and microsatellite analyses (see text for further explanation)
Four wheat microsatellites were analysed in the parents (Table 1). One of which, WMS6, has been located on 5RL in rye (homoeologous to Triticeae 4L; Börner et al. 1998, 1999) whereas the others are close to WMS6 on 4B in wheat (Röder et al. 1998). Only WMS6 was scored in the whole population, but was not significantly associated with plant height (Table 2). A few of the barley microsatellites that were screened were polymorphic in the parents, but none of them were polymorphic in the bulks (Table 1).
Previously, Rorat et al. (1991) sequenced the endosperm-specific β-amylase gene, β-amy1, from the beginning of exon 5. In the present study, this gene was sequenced from intron 3 to intron 6 in the DH parents. Four differences between the parents in exon 4 (EM-1: GenBank accession no. EU117383) were observed. Exon 4 of the endosperm-specific β-amylase gene differed in 35 nucleotides from the ubiquitous β-amylase gene sequenced by Sadowski et al. (1993). A SNP-BAMY marker was created based on one difference, and it showed a strong association with plant height (Tables 1, 2).
Genetic mapping
A QTL analysis was performed to determine distances between markers and the dwarfing gene. In total, 14 markers (microsatellites RMS1115, SCM151, SCM166, SCM268, WMS6, REMS1218; RAPDs APR5.2430, OPR-06780, OPB-18425, OPA-08845, P1471005; exon-intron-based marker ET2280; SNPs SNP-REMS1218, SNP-BAMY) were analysed in the mapping population. Afterwards, the results of REMS1218 and SNP-REMS1218 were combined (REMS1218com). The markers were arranged into two linkage groups which both contained markers known to be located on 5R: linkage group 1, consisting of RMS1115, SCM151, SCM166, SCM268, and OPB-18425; linkage group 2, containing WMS6, REMS1218com, SNP-BAMY, and ET2280. The RAPD markers APR5.2430, OPR-06780, OPA-08845, and P1471005 remained unlinked.
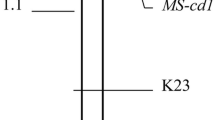
A major QTL (LOD score = 15.4) for plant height, which corresponds to Ddw1 was found on linkage group 2. The markers WMS6, REMS1218com, and SNP-BAMY were analysed also in the smaller population. The LOD score arose to 22.4 (variance-explained 67.8%) when the QTL analysis was performed with the pooled data from the two populations containing 158 individuals. The QTL was located between the markers WMS6 and REMS1218com, 13 cM from REMS1218 (Fig. 4).

A QTL likelihood map for plant height
Running a BLASTX search for the REMS1218 sequence (BE587316) against the EST database showed a significant genetic identity (E value 0.0, max identity 91–94%) with mRNAs for germin- and oxalate oxidase-like proteins in wheat and barley.
Discussion
In Finland, the reduction of straw height has been a major goal in rye breeding. It has been inconsistently achieved by incorporating the EM-1 mutant, which carries the dominant dwarfing gene, Ddw1 (Kobyliansky 1972), into the pedigree of rye breeding lines. The problem has been that it is impossible to get rid of the recessive allele for the tall growth habit, which has made it difficult to purify a breeding line at each of the various selection stages.
Four markers linked to plant height were found when almost 600 different primers or primer pairs were screened. The best marker, REMS1218com (Figs. 2, 3; Table 2), is a combination of the results from microsatellite and SNP analyses. Its recombinational distance from the gene yields a 13% misclassification. However, for a rye breeder the SNP-REMS1218 marker alone is an adequate tool identifying the desired individuals (plants homozygous for EM-1 allele) during breeding. It is the first efficient PCR-based marker linked to Ddw1 to be developed.
According to the genetic map of Korzun et al. (2001), the distance between WMS6 and Ddw1 on 5RL was 31.9 cM, which corresponds to the distance (30.2 cM) in the present study. However, the distance between SNP-BAMY and Ddw1 (36.6 cM) was about twice as long as the distance between the isozyme marker β-amy-R1 and Ddw1 in the map of Korzun et al. (2001). The markers in the other linkage group (RMS1115, SCM151, SCM166, SCM268, and OPB-18425) have previously been mapped to the centromere region of chromosome 5R, far from the linkage group containing Ddw1 on the distal end of the 5RL (data provided from BAZ, Germany; González et al. 2002; Hackauf and Wehling 2002a; Saal and Wricke 1999). A RAPD primer, APR5.2 has also been mapped both proximal and distal to Ddw1 on 5RL in rye (Bednarek et al. 2003; Masojć et al. 2001, Milczarski et al. 2007), but in the present study APR5.2430 showed no linkage to Ddw1.
Several factors hindered finding a closer marker to Ddw1. The ‘short’ bulk likely included both individuals homozygous for EM-1 allele and heterozygotes, which interfered with finding of marker alleles for the tall growth habit. In addition, the distal location of Ddw1 on chromosome 5RL increases the map distance relative to the physical distance in this region. Further, it has been observed in hexaploid wheat that the chromosome arm 4DL, which is homoeologous to the distal end of rye chromosome arm 5RL (Devos et al. 1993), has a very low level of polymorphism (Rodriguez Milla and Gustafson 2001).
According to the BLASTX search, the sequence REMS1218 (BE587316) has similarities with genes encoding germin-like proteins, which accumulate in germinating embryos in cereals. These proteins generate peroxide and have oxalate oxidase activity (e.g., Caliskan and Cuming 1998; Schweizer et al. 1999, Zimmermann et al. 2006). It has been suggested that germin-like oxalate oxidase (gl-OXO) restricts cell growth by participating in cell wall modification by producing hydrogen peroxide for cross-linking of cell wall components (Caliskan and Cuming 1998; Çalişkan 2000).
SNP markers are useful tools for various applications, including diagnostics, phylogenetic analyses, high-resolution maps, and mapping of traits (Rafalski 2002). Recently, SNP markers for short straw in oat have been generated for rapid screening of progenies in breeding programs (Tanhuanpää et al. 2006). Even though the SNP-REMS1218 marker developed in the current study is located quite far from the Ddw1 locus, in a cross-pollinating species MAS is the only means of purifying lines from the tall alleles. Shortening the straw can prevent lodging and crop damage for rye breeding generally.
References
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Becker J, Heun M (1995) Barley microsatellites: allele variation and mapping. Plant Mol Biol 27:835–845
Bednarek PT, Masojć P, Lewandowska R, Myśków B (2003) Saturating rye genetic map with amplified fragment length polymorphism (AFLP) and random amplified polymorphic DNA (RAPD) markers. J Appl Genet 44:21–33
Börner A, Plaschke J, Korzun V, Worland AJ (1996) The relationships between the dwarfing genes of wheat and rye. Euphytica 89:69–75
Börner A, Korzun V, Worland AJ (1998) Comparative genetic mapping of loci affecting plant height and development in cereals. Euphytica 100:245–248
Börner A, Korzun V, Voylokov AV, Weber WE (1999) Detection of quantitative trait loci on chromosome 5R of rye (Secale cereale L.). Theor Appl Genet 98:1087–1090
Çalişkan M (2000) Germin, an oxalate oxidase, has a function in many aspects of plant life. Turk J Biol 24:717–724
Caliskan M, Cuming AC (1998) Spatial specificity of H2O2-generating oxalate oxidase gene expression during wheat embryo germination. Plant J 15:165–171
Devos KM, Atkinson MD, Chinoy CN, Francis HA, Harcourt RL, Koebner RMD, Liu CJ, Masojć P, Xie DX, Gale MD (1993) Chromosomal rearrangements in the rye genome relative to that of wheat. Theor Appl Genet 85:673–680
Francia E, Tacconi G, Crosatti C, Barabaschi D, Bulgarelli D, Dall’Aglio E, Valè G (2005) Marker assisted selection in crop plants. Plant Cell Tissue Org Cult 82:317–342
González C, Camacho MV, Benito C (2002) Chromosomal location of 46 new RAPD markers in rye (Secale cereale L.). Genetica 115:205–211
Hackauf B, Wehling P (2002a) Development of microsatellite markers in rye: map construction. In: Osiński R (ed) Proceedings of the EUCARPIA Rye Meeting, July 4–7, 2001. Plant Breeding and Acclimatization Institute, Radzików, pp 333–340
Hackauf B, Wehling P (2002b) Identification of microsatellite polymorphisms in an expressed portion of the rye genome. Plant Breed 121:17–25
Haldane JBS, Smith CAB (1947) A new estimate for the linkage between the genes for colour-blindness and haemophilia in men. Ann Eugen 14:10–31
Khlestkina EK, Than MHM, Pestsova EG, Röder MS, Malyshev SV, Korzun V, Börner A (2004) Mapping of 99 new microsatellite-derived loci in rye (Secale cereale L.) including 39 expressed sequence tags. Theor Appl Genet 109:725–732
Kobyliansky VD (1972) On genetics of the dominant factor of short-strawed rye. Genetika 8:12–17
Korzun V, Melz G, Börner A (1996) RFLP mapping of the dwarfing (Ddw1) and hairy peduncle (Hp) genes on chromosome 5 of rye (Secale cereale L.). Theor Appl Genet 92:1073–1077
Korzun V, Röder M, Worland AJ, Börner A (1997) Intrachromosomal mapping of genes for dwarfing (Rht12) and vernalization response (Vrn1) in wheat by using RFLP and microsatellite markers. Plant Breed 116:227–232
Korzun V, Malyshev S, Voylokov AV, Börner A (2001) A genetic map of rye (Secale cereale L.) combining RFLP, isozyme, protein, microsatellite and gene loci. Theor Appl Genet 102:709–717
Lander ES, Green P, Abrahamson J, Barlow A, Daly M, Lincoln SE, Newburg L (1987) MAPMAKER: An interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1:174–181
Li G, Quiros CF (2001) Sequence-related amplified polymorphism (SRAP), a new marker system based on a simple PCR reaction: its application to mapping and gene tagging in Brassica. Theor Appl Genet 103:455–461
Liu Z-W, Biyashev RM, Saghai Maroof MA (1996) Development of simple sequence repeat DNA markers and their integration into a barley linkage map. Theor Appl Genet 93:869–876
Marshall HG, Murphy CF (1981) Inheritance of dwarfness in three oat crosses and relationship of height to panicle and culm length. Crop Sci 21:335–338
Masojć P, Myśków B, Milczarski P (2001) Extending a RFLP-based genetic map of rye using random amplified polymorphic DNA (RAPD) and isozyme markers. Theor Appl Genet 102:1273–1279
Michelmore RW, Paran I, Kesseli RV (1991) Identification of markers linked to disease-resistance genes by bulked segregant analysis: A rapid method to detect markers in specific genomic regions by using segregating populations. Proc Natl Acad Sci USA 88:9828–9832
Milach SCK, Federizzi LC (2001) Dwarfing genes in plant improvement. Adv Agron 73:35–63
Milach SCK, Rines HW, Phillips RL, Stuthman DD, Morikawa T (1998) Inheritance of a new dwarfing gene in oat. Crop Sci 38:356–360
Milczarski P, Banek-Tabor A, Lebiecka K, Stojałowski S, Myśków B, Masojć P (2007) New genetic map of rye composed of PCR-based molecular markers and its alignment with the reference map of the DS2 × RXL10 intercross. J Appl Genet 48:11–24
Oetting WS, Lee HK, Flanders DJ, Wiesner GL, Sellers TA, King RA (1995) Linkage analysis with multiplexed short tandem repeat polymorphisms using infrared fluorescence and M13 tailed primers. Genomics 30:450–458
Poulsen GB, Kahl G, Weising K (1993) Abundance and polymorphism of simple repetitive DNA sequences in Brassica napus L. Theor Appl Genet 85:994–1000
Rafalski JA (2002) Novel genetic mapping tools in plants: SNPs and LD-based approaches. Plant Sci 162:329–333
Rafalski A, Madej L, Wiśniewska I, Gaweł M (2002) The genetic diversity of components of rye hybrids. Cell Mol Biol Lett 7:471–475
Ramsay L, Macaulay M, degli Ivanissevich S, MacLean K, Cardle L, Fuller J, Edwards KJ, Tuvesson S, Morgante M, Massari A, Maestri E, Marmiroli N, Sjakste T, Ganal M, Powell W, Waugh R (2000) A simple sequence repeat-based linkage map of barley. Genetics 156:1997–2005
Röder MS, Plaschke J, König SU, Börner A, Sorrells ME, Tanksley SD, Ganal MW (1995) Abundance, variability and chromosomal location of microsatellites in wheat. Mol Gen Genet 246:327–333
Röder MS, Korzun V, Wendehake K, Plaschke J, Tixier M-H, Leroy P, Ganal MW (1998) A microsatellite map of wheat. Genetics 149:2007–2023
Rodriguez Milla MA, Gustafson JP (2001) Genetic and physical characterization of chromosome 4DL in wheat. Genome 44:883–892
Rorat T, Sadowski J, Grellet F, Daussant J, Delseny M (1991) Characterization of cDNA clones for rye endosperm β-amylase and analysis of β-amylase deficiency in rye mutant lines. Theor Appl Genet 83:257–263
Rozen S, Skaletsky HJ (2000) Primer3 on the WWW for general users and for biologist programmers. In: Krawetz S, Misener S (eds) Bioinformatics methods and protocols: methods in molecular biology. Humana Press, Totowa, pp 365–386. Source code available at http://fokker.wi.mit.edu/primer3/
Saal B, Wricke G (1999) Development of simple sequence repeat markers in rye (Secale cereale L.). Genome 42:964–972
Sadowski J, Rorat T, Cooke R, Delseny M (1993) Nucleotide sequence of a cDNA clone encoding ubiquitous β-amylase in rye (Secale cereale L.). Plant Physiol 102:315–316
Schweizer P, Christoffel A, Dudler R (1999) Transient expression of members of the germin-like gene family in epidermal cells of wheat confers disease resistance. Plant J 20:541–552
Sturm W, Engel K-H (1980) Trisome analysis of Hl allel of short strawiness in Secale cereale L. Arch Züchtungsforsch 10:31–35
Tanhuanpää P, Kalendar R, Laurila J, Schulman AH, Manninen O, Kiviharju E (2006) Generation of SNP markers for short straw in oat (Avena sativa L.). Genome 49:282–287
Tenhola-Roininen T, Tanhuanpää P, Immonen S (2005) The effect of cold and heat treatments on the anther culture response of diverse rye genotypes. Euphytica 145:1–9
Tenhola-Roininen T, Immonen S, Tanhuanpää P (2006) Rye doubled haploids as a research and breeding tool—a practical point of view. Plant Breed 125:584–590
Zimmermann G, Bäumlein H, Mock H-P, Himmelbach A, Schweizer P (2006) The multigene family encoding germin-like proteins of barley. Regulation and function in basal host resistance. Plant Physiol 142:181–192
Acknowledgments
We thank Marja-Riitta Arajärvi, Leena Ramstedt, and Sirpa Moisander for their efficient technical assistance in DNA marker analyses. Anneli Virta and Johanna Rusi are warmly thanked for running DNA markers with the MegaBACE and for DNA sequencing. We thank Maria Erkkilä for the barley β-amylase primers. Boreal Plant Breeding Ltd. kindly provided all the rye material used in this study. Professor Alan Schulman is acknowledged for the language revision of the manuscript. The Finnish Ministry of Agriculture and Forestry, Heikki and Hilma Honkanen Foundation, the Finnish Academy of Sciences (project number 112053), and Finnish Cultural Foundation are gratefully acknowledged for financially supporting this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tenhola-Roininen, T., Tanhuanpää, P. Tagging the dwarfing gene Ddw1 in a rye population derived from doubled haploid parents. Euphytica 172, 303–312 (2010). https://doi.org/10.1007/s10681-009-9982-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10681-009-9982-8