
Abstract
More information is required to improve our understanding of the presence of organic ultraviolet absorbents (UVAs) in organisms and the risks posed to biota. Effective removal of lipids in the determination of UVAs in biological samples is a sample-processing bottleneck. In this study, we optimized a multistep purification method for fish tissue samples extracted using an ultrasonic-assisted extraction method. The purification method involved performing Florisil column chromatography, redissolving the extract in ethanol, and then performing dispersive solid-phase extraction using primary and secondary amine, Florisil, and C18 silica sorbents. The purified samples were analyzed by gas chromatography–mass spectrometry. The method was effective and reliable, and was used to detect trace concentrations (ng/g) of 12 UVAs in fish tissue samples. The mean recovery range of the UVAs in fish tissue extracts was 65.4–118%, and the method detection limit range was 0.20–2.50 ng/g dw. The validated method was used to analyze 12 UVAs in fish samples from a local supermarket.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Exposure to ultraviolet (UV) radiation during outdoor activities increases the risk for skin cancer (Fent et al. 2008). To reduce this risk, personal care products containing organic ultraviolet absorbents (UVAs) are widely used and have been produced in large quantities (Li et al. 2007; Moehrle 2008; Richardson 2009). UVAs are also used to prevent aging and the degradation of industrial products (Coltro et al. 2003). Because of their long-term and extensive use, UV filter chemicals are frequently found in water (Poiger et al. 2004), sediment (Plagellat et al. 2006), dust (Wang et al. 2013), and tissue (Nagtegaal et al. 1997; Fent et al. 2010; Bachelot et al. 2012) samples. In humans, they have been found in milk, blood, and urine (Zhang et al. 2013; Rodríguez-Gómez et al. 2015). UVAs can disrupt the endocrine system and cause developmental toxicity (Coronado et al. 2008; Fent et al. 2008; Blüthgen et al. 2012), and some UVAs are suspected to be persistent in the environment (Kameda et al. 2007; Lai et al. 2014). Many UVAs are strongly lipophilic and may bioaccumulate (Gago-Ferrero et al. 2015). Public interest in the toxicities of organic UVAs and their environmental risks is increasing. Further studies are required to improve our understanding of the presence of organic UVAs in environmental media and the risks posed to biota.
Soxhlet extraction, microwave-assisted extraction, pressurized liquid extraction, and solid–liquid extraction methods have been developed and applied to detect UVAs in biological samples (Gago-Ferrero et al. 2012). During extraction, a lipid fraction can be co-extracted and may cause strong interference in instrumental analysis. Consequently, lipid removal before analysis is indispensable. Several purification methods, such as gel permeation chromatography, solid-phase extraction, and reversed-phase high-performance liquid chromatography, have been applied for lipid removal in the process of UVA detection (Balmer et al. 2005; Buser et al. 2006; Meinerling and Daniels 2006; Zenker et al. 2008; Mottaleb et al. 2009; Fent et al. 2010; Sang and Leung 2016). However, these procedures are time-consuming and require large solvent volumes (He et al. 2017).
Recently, dispersive solid-phase extraction (d-SPE) was developed and applied to remove co-extracted materials (e.g., lipids and proteins) using primary and secondary amine (PSA) and C18 silica as sorbents for UVAs in fish muscle, human placenta, and other tissues (Berlioz-Barbier et al. 2014; Groz et al. 2014; He et al. 2017; Vela-Soria et al. 2017). In some previous studies, good purification was achieved using QuEChERS with acetonitrile, which lipids are poorly soluble in, as the extractant. However, acetonitrile could be ineffective for the extraction of some strongly lipophilic chemicals (Anastassiades et al. 2003; Norli et al. 2011; Satpathy et al. 2011; Groz et al. 2014). Groz et al. (2014) reported that the recovery of bumetrizole (UV-326) was less than 50% in the detection of UVAs in marine mussels. To extract lipophilic UVAs, some semi-polar solvents may be more suitable for use as extractants. Optimization of d-SPE for UVAs is necessary.
An effective pretreatment method is a prerequisite for accurate analysis of UVAs in biological samples. However, cheap and efficient pretreatment methods for the simultaneous determination of polar, semi-polar, and lipophilic UVAs in biota samples have rarely been reported. The aim of this study was to develop a cheap and sensitive method for detecting trace amounts of multiclass UVAs in biological samples. We selected the 12 most common UVAs in cosmetics and industrial products as the targets. The UVAs were extracted by ultrasonic-assisted extraction (UAE), purified by a combination of column fractionation, redissolution, and d-SPE, and finally determined by gas chromatography–mass spectrometry (GC–MS). The developed method was validated and applied to detect UVAs in fish muscle and liver samples.
Materials and methods
Chemicals and reagents
Standards of 2-(2′-hydroxy-3′,5′-di-tert-butylphenyl)-5-chlorobenzotriazole (UV-327) and octrizole (UV-329) were purchased from Tokyo Chemical Industry (Tokyo, Japan). Benzophenone (BP) was obtained from Chem Service (West Chester, PA, USA). Octocrylene was purchased from Sigma-Aldrich (St. Louis, MO, USA). UV-326 and deuterated BP-d10 were purchased from Chiron (Trondheim, Norway). 2-(2′-Hydroxy-3′,5′-di-tert-butylphenyl) benzotriazole (UV-320) was purchased from Dr. Ehrenstorfer (Augsburg, Germany). Deuterated phenanthrene-d10, 2-hydroxy-4-methoxybenzophenone, 2-ethylhexyl-4-methoxycinnamate, homosalate, ethylhexyl salicylate, octyl dimethyl p-aminobenzoic acid, and 3-(4-methylbenzylidene) camphor were obtained from AccuStandard (New Haven, CT, USA). Their physicochemical properties had been reported in detail elsewhere (Tang et al. 2019).
Stock solutions of the 12 analytes and two deuterated surrogates were prepared individually in n-hexane. Working solutions for the 12 analytes were prepared at 10 mg/L. All of the standard solutions were stored in amber glass containers in the dark at − 20 °C.
Chromatographic grade solvents (acetone, acetonitrile, dichloromethane, ethanol, formic acid, n-hexane, and methanol) were obtained from J.T. Baker (Phillipsburg, PA, USA). Analytical grade anhydrous sodium sulfate (Na2SO4), aluminum oxide (100–200 mesh), silica gel (100–200 mesh), and Florisil (60–100 mesh and 100–200 mesh) were all purchased from Sinopharm Chemical Reagent Co. (Beijing, China). PSA sorbent (Bondesil-PSA) and Bondesil-C18 (ø 40 μm) were purchased from Agilent Technologies (Santa Clara, CA, USA).
Sample preparation
Samples of carp (Cyprinus carpio) and black carp (Mylopharyngodon piceus) were purchased from a local supermarket. Each fish was carefully skinned and dissected with a stainless steel scalpel. The dorsal muscles and livers were analyzed, and all other tissues were discarded. Each sample was freeze-dried, ground, homogenized, and then stored at − 20 °C until analysis.
A 4-g sample of freeze-dried homogenized muscle or 2 g of liver was mixed with 15 mL of acetone/n-hexane (1:1, v/v) and then sonicated in an ultrasonic bath (40 kHz) for 15 min. The sample was then centrifuged at 5000 rpm for 5 min, and the clear supernatant was collected in a glass tube. The extraction process was carried out twice, and the extracts were combined.
The extract was transferred to a 10-mm i.d. glass column containing 4 cm of Na2SO4 and 4 cm of Florisil, and the filtrate was collected. A 15 mL aliquot of the filtrate was evaporated to dryness using a rotatory evaporator, and the residue was redissolved in 2 mL of ethanol. The extract was transferred to a glass centrifuge tube containing a mixture of sorbents (200 mg of C18, 50 mg of PSA, and 300 mg of Florisil (100–200 mesh)), shaken for more than 1 min, and then centrifuged for 5 min. An aliquot (1 mL) of the supernatant was evaporated to dryness and then diluted to a final volume of 0.5 mL with n-hexane. Internal standards were added to the extract before analysis by GC–MS.
Instrumental analysis
The extracts were analyzed using a gas chromatograph (7890, Agilent Technologies) coupled with a mass spectrometer (5975, Agilent Technologies). The column was a DB-5MS column (30 m × 0.25 μm, 0.25-μm film thickness, Agilent Technologies). The gas chromatograph oven temperature was held at 80 °C for 2 min, increased at 10 °C/min to 300 °C, and held at 300 °C for 15 min. The injector temperature was 250 °C. The flow rate of the helium carrier gas was 1 mL/min. For analysis, 1 μL aliquots of the samples were injected in splitless mode. The mass detector was operated in electron impact ionization mode using an electron energy of 70 eV. The ionization source was kept at 230 °C. The interface temperature was 280 °C, and the quadrupole temperature was 150 °C. The retention times and quantification/confirmation ions in each case are presented in detail elsewhere (Tang et al. 2019).
Method validation
The linearity of the method was evaluated by analyzing UVA standards at six concentrations between 10 and 400 μg/L. The instrument detection limit (IDL) range with a signal-to-noise ratio of 3:1 was 0.33–4.90 μg/L. Recovery tests were performed using three types of fish tissue with mixtures of standard solutions at 50 and 100 ng/g. The matrix effect (ME) was calculated using ME = (Ais − Aiu) / Ai (Gómez et al. 2006), where Ais is the analyte peak area corresponding to each point from the calibration curve for the spiked fish tissue extract, Aiu is the analyte peak area in the unspiked fish tissue extract, and Ai is the analyte peak area in the pure solvent at the same concentration as the corresponding spiked fish tissue extract. The method detection limits (MDLs) were defined as the minimum concentrations for detection of the analytes based on the instrument detection limits and MEs from spiked fish tissue extracts.
The standard solution and n-hexane were detected in each batch of seven samples to evaluate procedural contamination and instrument performance, respectively. To prevent contamination, personnel conducting the experiments were not allowed to use cosmetics or other skincare products likely containing UVAs. Glass, stainless steel, and polytetrafluoroethylene laboratory equipment were cleaned three times with acetone and n-hexane before use.
Results and discussion
Optimization of extractant
We investigated the use of a weak polar solvent or mixture of solvents as the extracting agent for UVAs with varying lipophilicity (log Kow = 3–7). A mixture of n-hexane and acetone reportedly gave satisfactory extraction efficiencies as an extract in a previous study (Kim et al. 2011). In the determination of UVAs in fish, satisfactory extraction efficiencies have been reported for ultrasonic-assisted extraction with methanol (Peng et al. 2015). Therefore, we used a mixture of acetone/n-hexane (1:1, v/v) and methanol to extract the UVAs from fish samples.
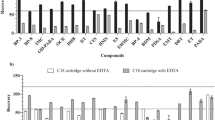
First, we spiked fish muscle samples with the target analytes and extracted them with methanol and acetone/n-hexane. Next, we separated the supernatant by centrifugation. The clear supernatant (2 mL) was added to a tube containing 100 mg of PSA sorbent and 100 mg of C18. After shaking and centrifuging, 0.5 mL of the supernatant was removed and evaporated to dryness under a gentle stream of nitrogen. The residue was dissolved in 0.5 mL of n-hexane before GC–MS analysis. For most of the UVAs, better recoveries were obtained with acetone/n-hexane than methanol (Fig. 1). In the methanol extraction, a precipitate appeared in the solution after redissolution of the residue, which may have contributed to the relatively low recoveries. Therefore, we selected acetone/n-hexane (1:1, v/v) as the extractant.

Recoveries (%) of the 12 organic ultraviolet absorbents (UVAs) from fish muscle with different extraction solvents
We also investigated the number of extraction cycles. The samples were extracted for 15 min using 15 mL of acetone and n-hexane (1:1, v/v), and this process was repeated three times. The extracts from each of the three extractions were kept separated for analysis. In the third extract, hardly any compound was detected. The use of fewer extraction cycles can save time and reduce consumption of solvents. Therefore, we selected 2 cycles for the optimized method.
Purification
To detect contaminants present at trace levels, extracts usually have to be concentrated. In a preexperiment, we tested d-SPE to purify the concentrated ultrasonication extracts. However, d-SPE alone was unable to remove all impurities effectively because the adsorption capacity of the adsorbent was limited and there was an excess of co-extracted materials in concentrated extracts from ultrasonic-assisted extraction with acetone and n-hexane. To give satisfactory cleanup, more sorbent needs to be used in d-SPE, but this increases the cost and may affect the recoveries of the analytes. Accordingly, the content of co-extracted materials needs to be reduced before d-SPE. The sorbents alumina, silica gel, and Florisil are commonly used to remove polar interfering substances in the detection of trace organic pollutants (Buser et al. 2006; Nakata et al. 2009, 2012; Bernsmann et al. 2014; Chang et al. 2014). In addition, some weakly polar or non-polar interferences can be eliminated by redissolution in appropriate solvents. In this study, we used a three-stage process of column fractionation, redissolution, and d-SPE to clean the sample extracts.
The most appropriate sorbents (alumina, silica gel, and Florisil [60–100 and 100–200 mesh]) for use in the chromatography column were selected by performing batch tests. Details are given in the Supplementary Material. The Florisil (60–100 mesh) had poor adsorption for most target analytes in the standard solution (see Supplementary Material: Table S1). Therefore, when used as a sorbent, Florisil (60–100 mesh) both removes some polar interfering substances and has low adsorption of the target chemicals. The elution step may be omitted in real sample pretreatment. We also investigated the quantity of sorbent added to the chromatography column (Supplementary Material). The results showed that purification with a column containing 4 cm of Florisil (60–100 mesh) and 4 cm of Na2SO4 gave clear chromatographic peaks (see Supplementary Material: Fig. S1).
For redissolution of the residues, we screened ethanol, acetonitrile, methanol, and a mixture of ethanol and acetonitrile (1:1, v/v) in batch tests. Details are given in the Supplementary Material. The recoveries of the 12 UVAs with the different solvents are shown in Fig. 2. Compared with other solvents, ethanol gave acceptable recoveries for the UVAs, especially for UV-320 and UV-326. This may be because ethanol is less polar than the other solvents, or it affects the adsorptivity of the sorbent in d-SPE. After our comparison of the different solvents, we selected ethanol as the most appropriate solvent for redissolution.

Recoveries of the organic ultraviolet absorbents (UVAs) when extracts were redissolved in different solvents
We performed tests to optimize the d-SPE process to ensure that as many interfering substances were removed as possible. We determined the recoveries of the UVAs after extract purification using PSA and C18 sorbents. Details are given in the Supplementary Material. The best UVA recoveries were achieved using 200–300 mg of the C18 sorbent and 100 mg of the PSA sorbent (see Supplementary Material: Table S2). However, interfering peaks were found in the chromatograms when only 100 mg of the PSA sorbent was used. Therefore, the quantities of the sorbents were further optimized to achieve the best cleanup for different types of samples, which likely have different MEs. We tested three sorbent combinations (see Supplementary Material: Table S3). Using the group 1 sorbent combination (200 mg C18 + 500 mg Florisil [100–200 mesh]), 2-hydroxy-4-methoxybenzophenone and 3-(4-methylbenzylidene) camphor could not be quantitatively analyzed because of interferences in the chromatograms. By contrast, the interfering peaks were smaller, and the UVA recoveries were satisfactory with group 2 (200 mg C18 + 100 mg PSA + 150 mg Florisil [100–200 mesh]) and group 3 (200 mg C18 + 50 mg PSA + 300 mg Florisil [100–200 mesh]) sorbent combinations. The group 3 sorbent contained less PSA than the group 2 sorbent, and we selected group 3 as the most appropriate sorbent for the d-SPE process to remove substances that could interfere with the analysis of UVAs in fish tissues.
Method validation
The analytical performance data are shown in Table 1. We evaluated the recoveries of the optimized method by analyzing fish muscle and liver samples spiked with UVAs at two levels. The recoveries of most of analytes in both muscle and liver samples were ≥ 70%. Lower recoveries were obtained for some analytes in liver samples, and were likely caused by the higher lipid content and more complex composition of the extract compared with those from the other tissues. The relative standard deviations of the analyte concentrations in carp muscle and black carp muscle samples were ≤ 7.9% and ≤ 16.4%, respectively. The relative standard deviations of the concentrations of all the analytes, except octyl dimethyl p-aminobenzoic acid (21.0%), in the liver samples were ≤ 16.1%. Overall, the method gave very reproducible results when used to simultaneously determine the 12 organic UVAs in fish tissue samples. With 4 g of sample, the MDL range was 0.2–2.5 ng/g dw and the ME range was − 13.1–15.5% (Table 1). These results are satisfactory for determination of trace UVAs in complex biological matrices.
Analysis of fish samples
The established method was used to analyze fish samples collected from a local supermarket. The mean UVA concentrations in carp muscle, black carp muscle, and mixed liver samples are shown in Fig. 3. The concentrations of BP, UV-326, ethylhexyl salicylate, 2-ethylhexyl-4-methoxycinnamate, UV-320, and octocrylene were lower than the corresponding MDLs in some samples. The total concentrations of the 12 organic UVAs in the carp muscle, black carp muscle, and liver samples were 38.8, 19.3, and 59.9 ng/g dw, respectively.

Concentrations of organic ultraviolet absorbents (UVAs) in the fish muscle and fish liver samples
Conclusions
Simple and sensitive analytical methods are necessary to detect and analyze the distributions of emerging contaminants in environmental samples. Sample preparation is a key step for analysis at trace levels, and interferences introduced during pretreatment can cause serious deviations in the results. In this work, we developed a method for simultaneously determining several groups of organic UVAs in fish tissue samples. This method fulfills the validity criteria for trace analysis and was successfully applied to quantification of UVAs in fish samples collected from a local supermarket.
References
Anastassiades, M., Lehotay, S. J., Štajnbaher, D., & Schenck, F. J. (2003). Fast and easy multiresidue method employing acetonitrile extraction/partitioning and “dispersive solid-phase extraction” for the determination of pesticide residues in produce. Journal of AOAC International, 86(2), 412–431.
Bachelot, M., Li, Z., Munaron, D., Le Gall, P., Casellas, C., Fenet, H., & Gomez, E. (2012). Organic UV filter concentrations in marine mussels from French coastal regions. Science of the Total Environment, 420(6), 273–279. https://doi.org/10.1016/j.scitotenv.2011.12.051.
Balmer, M. E., Buser, H. R., Müller, M. D., & Poiger, T. (2005). Occurrence of some organic UV filters in wastewater, in surface waters, and in fish from Swiss lakes. Environmental Science and Technology, 39(4), 953–962. https://doi.org/10.1021/es040055r.
Berlioz-Barbier, A., Buleté, A., Faburé, J., Garric, J., Cren-Olivé, C., & Vulliet, E. (2014). Multi-residue analysis of emerging pollutants in benthic invertebrates by modified micro-quick-easy-cheap-efficient-rugged-safe extraction and nanoliquid chromatography–nanospray–tandem mass spectrometry analysis. Journal of Chromatography A, 1367, 16–32. https://doi.org/10.1016/j.chroma.2014.09.044.
Bernsmann, T., Albrecht, M., & Fürst, P. (2014). Fast sample preparation for routine determination of PCDD/F, PCB and PBDE in food and feed. Organohalogen Compound, 76, 1281–1284.
Blüthgen, N., Zucchi, S., & Fent, K. (2012). Effects of the UV filter benzophenone-3 (oxybenzone) at low concentrations in zebrafish (Danio rerio). Toxicology and Applied Pharmacology, 263(2), 184–194. https://doi.org/10.1016/j.taap.2012.06.008.
Buser, H. R., Balmer, M. E., Schmid, P., & Kohler, M. (2006). Occurrence of UV filters 4-methylbenzylidene camphor and octocrylene in fish from various Swiss rivers with inputs from wastewater treatment plants. Environmental Science and Technology, 40(5), 1427–1431. https://doi.org/10.1021/es052088s.
Chang, Y., Lee, W., Wang, L., Yang, H., Cheng, M., Lu, J., Tsai, Y., & Young, L. (2014). Effects of waste cooking oil-based biodiesel on the toxic organic pollutant emissions from a diesel engine. Applied Energy, 113(113), 631–638. https://doi.org/10.1016/j.apenergy.2013.08.005.
Coltro, L., Padula, M., Saron, E. S., Borghetti, J., & Buratin, A. E. P. (2003). Evaluation of a UV absorber added to PET bottles for edible oil packaging. Packaging Technology and Science, 16(1), 15–20. https://doi.org/10.1002/pts.607.
Coronado, M., De Haro, H., Deng, X., Rempel, M. A., Lavado, R., & Schlenk, D. (2008). Estrogenic activity and reproductive effects of the UV-filter oxybenzone (2-hydroxy-4-methoxyphenyl-methanone) in fish. Aquatic Toxicology, 90(3), 182–187. https://doi.org/10.1016/j.aquatox.2008.08.018.
Fent, K., Kunz, P. Y., & Gomez, E. (2008). UV filters in the aquatic environment induce hormonal effects and affect fertility and reproduction in fish. Chimia, 62(5), 368–375. https://doi.org/10.2533/chimia.2008.368.
Fent, K., Zenker, A., & Rapp, M. (2010). Widespread occurrence of estrogenic UV-filters in aquatic ecosystems in Switzerland. Environmental Pollution, 158(5), 1817–1824. https://doi.org/10.1016/j.envpol.2009.11.005.
Gago-Ferrero, P., Diaz-Cruz, M. S., & Barceló, D. (2012). An overview of UV-absorbing compounds (organic UV filters) in aquatic biota. Analytical and Bioanalytical Chemistry, 404(9), 2597–2610. https://doi.org/10.1007/s00216-012-6067-7.
Gago-Ferrero, P., Díaz-Cruz, M. S., & Barceló, D. (2015). UV filters bioaccumulation in fish from Iberian river basins. Science of the Total Environment, 518, 518–525. https://doi.org/10.1016/j.scitotenv.2015.03.026.
Gómez, M. J., Petrović, M., Fernández-Alba, A. R., & Barceló, D. (2006). Determination of pharmaceuticals of various therapeutic classes by solid-phase extraction and liquid chromatography-tandem mass spectrometry analysis in hospital effluent wastewaters. Journal of Chromatography A, 1114(2), 224–233. https://doi.org/10.1016/j.chroma.2006.02.038.
Groz, M. P., Bueno, M. M., Rosain, D., Fenet, H., Casellas, C., Pereira, C., Maria, V., Bebianno, M. J., & Gomez, E. (2014). Detection of emerging contaminants (UV filters, UV stabilizers and musks) in marine mussels from Portuguese coast by QuEChERS extraction and GC–MS/MS. Science of the Total Environment, 493, 162–169. https://doi.org/10.1016/j.scitotenv.2014.05.062.
He, K., Timm, A., & Blaney, L. (2017). Simultaneous determination of UV-filters and estrogens in aquatic invertebrates by modified quick, easy, cheap, effective, rugged, and safe extraction and liquid chromatography tandem mass spectrometry. Journal of Chromatography A, 1509, 91–101. https://doi.org/10.1016/j.chroma.2017.06.039.
Kameda, Y., Tamada, M., Kanai, Y., & Masunaga, S. (2007). Occurrence of organic UV filters in surface waters, sediments and core sediments in Tokyo bay–organic UV filters are new POPs? Organohalogen Compound, 69, 263–266.
Kim, J. W., Ramaswamy, B. R., Chang, K., Isobe, T., & Tanabe, S. (2011). Multiresidue analytical method for the determination of antimicrobials, preservatives, benzotriazole UV stabilizers, flame retardants and plasticizers in fish using ultra high performance liquid chromatography coupled with tandem mass spectrometry. Journal of Chromatography A, 1218, 3511–3520. https://doi.org/10.1016/j.chroma.2011.04.006.
Lai, H., Ying, G., Ma, Y., Chen, Z., Chen, F., & Liu, Y. (2014). Field dissipation and plant uptake of benzotriazole ultraviolet stabilizers in biosolid-amended soils. Environmental Science: Processes & Impacts, 16(3), 558–566. https://doi.org/10.1039/C3EM00568B.
Li, W., Ma, Y., Guo, C., Hu, W., Liu, K., Wang, Y., & Zhu, T. (2007). Occurrence and behavior of four of the most used sunscreen UV filters in a wastewater reclamation plant. Water Research, 41(15), 3506–3512. https://doi.org/10.1016/j.watres.2007.05.039.
Meinerling, M., & Daniels, M. (2006). A validated method for the determination of traces of UV filters in fish using LC–MS/MS. Analytical and Bioanalytical Chemistry, 386(5), 1465–1473. https://doi.org/10.1007/s00216-006-0706-9.
Moehrle, M. (2008). Outdoor sports and skin cancer. Clinics in Dermatology, 26(1), 12–15. https://doi.org/10.1016/j.clindermatol.2007.10.001.
Mottaleb, M. A., Usenko, S., O’Donnell, J. G., Ramirez, A. J., Brooks, B. W., & Chambliss, C. K. (2009). Gas chromatography–mass spectrometry screening methods for select UV filters, synthetic musks, alkylphenols, an antimicrobial agent, and an insect repellent in fish. Journal of Chromatography A, 1216(5), 815–823. https://doi.org/10.1016/j.chroma.2008.11.072.
Nagtegaal, M., Ternes, T. A., Baumann, W., & Nagel, R. (1997). UV-filtersubstanzen in wasser und fischen. Umweltchem Okotox, 9(2), 79–86. https://doi.org/10.1007/BF02937843.
Nakata, H., Murata, S., & Filatreau, J. (2009). Occurrence and concentrations of benzotriazole UV stabilizers in marine organisms and sediments from the Ariake Sea, Japan. Environmental Science and Technology, 43(18), 6920–6926. https://doi.org/10.1021/es900939j.
Nakata, H., Shinohara, R., Nakazawa, Y., Isobe, T., Sudaryanto, A., Subramanian, A., Tanabe, S., Zakaria, M. P., Zheng, G. J., Lam, P. K. S., Kim, E. Y., Min, B. Y., We, S. U., Viet, P. H., Tana, T. S., Prudente, M., Frank, D., Lauenstein, G., & Kannan, K. (2012). Asia-Pacific mussel watch for emerging pollutants: distribution of synthetic musks and benzotriazole UV stabilizers in Asian and US coastal waters. Marine Pollution Bulletin, 64(10), 2211–2218. https://doi.org/10.1016/j.marpolbul.2012.07.049.
Norli, H. R., Christiansen, A., & Deribe, E. (2011). Application of QuEChERS method for extraction of selected persistent organic pollutants in fish tissue and analysis by gas chromatography mass spectrometry. Journal of Chromatography A, 1218(41), 7234–7241. https://doi.org/10.1016/j.chroma.2011.08.050.
Peng, X., Jin, J., Wang, C., Ou, W., & Tang, C. (2015). Multi-target determination of organic ultraviolet absorbents in organism tissues by ultrasonic assisted extraction and ultra-high performance liquid chromatography–tandem mass spectrometry. Journal of Chromatography A, 1384, 97–106. https://doi.org/10.1016/j.chroma.2015.01.051.
Plagellat, C., Kupper, T., Furrer, R., De Alencastro, L. F., Grandjean, D., & Tarradellas, J. (2006). Concentrations and specific loads of UV filters in sewage sludge originating from a monitoring network in Switzerland. Chemosphere, 62(6), 915–925. https://doi.org/10.1016/j.chemosphere.2005.05.024.
Poiger, T., Buser, H. R., Balmer, M. E., Bergqvist, P. A., & Müller, M. D. (2004). Occurrence of UV filter compounds from sunscreens in surface waters: regional mass balance in two Swiss lakes. Chemosphere, 55(7), 951–963. https://doi.org/10.1016/j.chemosphere.2004.01.012.
Richardson, S. D. (2009). Water analysis: emerging contaminants and current issues. Analytical Chemistry, 81(12), 4645–4677. https://doi.org/10.1021/ac9008012.
Rodríguez-Gómez, R., Zafra-Gómez, A., Dorival-García, N., Ballesteros, O., & Navalón, A. (2015). Determination of benzophenone-UV filters in human milk samples using ultrasound-assisted extraction and clean-up with dispersive sorbents followed by UHPLC–MS/MS analysis. Talanta, 134, 657–664. https://doi.org/10.1016/j.talanta.2014.12.004.
Sang, Z., & Leung, K. S. Y. (2016). Environmental occurrence and ecological risk assessment of organic UV filters in marine organisms from Hong Kong coastal waters. Science of the Total Environment, 566, 489–498. https://doi.org/10.1016/j.scitotenv.2016.05.120.
Satpathy, G., Tyagi, Y. K., & Gupta, R. K. (2011). A novel optimised and validated method for analysis of multi-residues of pesticides in fruits and vegetables by microwave-assisted extraction (MAE)–dispersive solid-phase extraction (d-SPE)–retention time locked (RTL)–gas chromatography–mass spectrometry with deconvolution reporting software (DRS). Food Chemistry, 127(3), 1300–1308. https://doi.org/10.1016/j.foodchem.2011.01.087.
Tang, Z., Zhong, F., Cheng, J., Nie, Z., Han, X., Han, Y., & Yang, Y. (2019). Concentrations and tissue-specific distributions of organic ultraviolet absorbents in wild fish from a large subtropical lake in China. Science of the Total Environment, 647, 1305–1313. https://doi.org/10.1016/j.scitotenv.2018.08.117.
Vela-Soria, F., Gallardo-Torres, M. E., Ballesteros, O., Díaz, C., Pérez, J., Navalón, A., Fernándezade, M. F., & Olea, N. (2017). Assessment of parabens and ultraviolet filters in human placenta tissue by ultrasound-assisted extraction and ultra-high performance liquid chromatography-tandem mass spectrometry. Journal of Chromatography A, 1487, 153–161. https://doi.org/10.1016/j.chroma.2017.01.041.
Wang, L., Asimakopoulos, A. G., Moon, H. B., Nakata, H., & Kannan, K. (2013). Benzotriazole, benzothiazole, and benzophenone compounds in indoor dust from the United States and East Asian countries. Environmental Science and Technology, 47(9), 4752–4759. https://doi.org/10.1021/es305000d.
Zenker, A., Schmutz, H., & Fent, K. (2008). Simultaneous trace determination of nine organic UV-absorbing compounds (UV filters) in environmental samples. Journal of Chromatography A, 1202, 64–74. https://doi.org/10.1016/j.chroma.2008.06.041.
Zhang, T., Sun, H., Qin, X., Wu, Q., Zhang, Y., Ma, J., & Kannan, K. (2013). Benzophenone-type UV filters in urine and blood from children, adults, and pregnant women in China: partitioning between blood and urine as well as maternal and fetal cord blood. Science of the Total Environment, 461(7), 49–55. https://doi.org/10.1016/j.scitotenv.2013.04.074.
Funding
This research was supported by the National Natural Science Foundation of China (Grant Nos. 41571445; 41877467) and the Fundamental Research Funds for the Central Universities (Grant No. 15MS60).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary data associated with this article can be found, in the online version, at
ESM 1
(DOCX 203 kb)
Rights and permissions
About this article

Cite this article
Zhong, F., Zhang, X., Li, G. et al. A new multistep purification method for simultaneously determining organic ultraviolet absorbents in fish tissue. Environ Monit Assess 191, 16 (2019). https://doi.org/10.1007/s10661-018-7139-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10661-018-7139-8