
Abstract
A procedure is detailed for the selective analysis of trace aluminum by flame atomic absorption spectrophotometer coupled with off-line column separation and preconcentration. Chelating resin was synthesized by covalent functionalization of Amberlite XAD-16 by 2-(2-hydroxyphenyl) benzoxazole. The modified resin was characterized using FT-IR spectroscopy, energy dispersive x-ray analysis, elemental analysis, thermogravimetric analysis/differential thermal analysis, and minimum energy run. The optimum sorption was at pH 9 ± 0.1 with corresponding t 1/2 of only 7 min. Many competitive anions and cations studied did not interfere at all in the selective determination of Al(III), at the optimized conditions. The resin shows maximum sorption capacity of 21.58 mg g−1 and can be regenerated up to 75 cycles without any discernible capacity loss. The Langmuir isotherm model provides the better correlation of the experimental data (r 2 = 0.999) in comparison to Freundlich isotherm model, while the Scatchard analysis revealed homogeneous binding sites in the chelating resin. Analytical figures of merit were evaluated by accuracy (standard reference materials and recovery experiment), precision (RSD <5 %), and detection limit (2.8 μg L−1). The applicability was demonstrated by analysis of trace aluminum in biological, environmental, and food samples.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Atomic absorption spectrometry (AAS) and inductively coupled plasma–mass spectrometry (ICP-MS) techniques have excellent analytical sensitivity and are the methods of choice for the determination of ultra-trace level elements in various matrices. However, it would be naive to assume that all laboratories that are required to perform trace level estimations are equipped with such sophisticated and expensive instruments. Further, direct determination of metal ions at trace levels by such sophisticated analytical techniques is limited, not only due to insufficient sensitivity but also to matrix interference. To overcome these difficulties, researchers have described various procedures for sample pretreatment. These include liquid–liquid extraction (Carasek 2000), solid-phase extraction (Silva et al. 2004), and cloud point extraction (Paleologos et al. 2005). The separation and preconcentration step is employed prior to the trace metal quantification, as adjuncts to such techniques. Flame atomic absorption spectrophotometer (FAAS) has been widely used for its advantages of less spectral interferences by concomitants and relatively lower running costs when compared to expensive flameless more sophisticated techniques which are often more sensitive to interferences (Horng and Lin 1997; Ottaway and Pradhan 1973; Bergmann and Hardt 1979; Ejaz et al. 1981; Popova et al. 1991).
Solid-phase extraction offers the advantage of high sensitivity and versatility due to the possibility of performing a simultaneous enrichment step. Chelating resins are superior in selectivity to solvent extraction and ion exchange due to their triple functions (i.e., ion exchange, chelating ability, and physical adsorption) (Dutta and Das 2008). These materials are highly efficient in removing metal ions, and often provide the needed selectivity based on the chelating moiety with an easy on-demand release and reusability. Simple, rapid, and economic solid-phase extraction methods have been developed and applied successfully for the estimation of trace levels of some metal ions in different environmental and biological samples from researcher’s laboratory (Islam et al. 2011a; Islam et al. 2012a; Islam et al. 2012b; Islam et al. 2011b; Islam et al. 2013a).
The continually improving technology for the extraction of aluminum from its biologically inert ores accelerates the exposure of non-essential biologically reactive aluminum to human ecology (Exley et al. 2009). Aluminum, which was previously considered benign, has been recently found to be a dementing ion and cause both bone and neurological disorders in humans. The potential toxicity of aluminum to humans and the environment has drawn the attention of many analysts in determination of very low aluminum concentrations (Erdemoglu et al. 2000; Neal et al. 2011; Matus and Kubova 2006).
Many 2-(2-hydroxyphenyl)-benzoxazole (HPB) complexes with aluminum (Hoveyda et al. 1993), gallium (Hoveyda et al. 1993), zinc (Henary and Fahrni 2002; Tian et al. 2008), copper (Zhang et al. 2006), and lithium (Qin et al. 2001) have been investigated so far in aqueous phase. According to the best of our literature knowledge, metal ions complexation with HPB in solid-phase extraction has never been reported. Herein, we report the synthesis and application of a novel aluminum-selective chelating resin by immobilizing HPB on Amberlite XAD-16 through covalent bonding, which tends to be more chemically stable to acidic eluents than those resins prepared by impregnation. This XAD-HPB resin was then characterized and systematically explored for its application in the separation/preconcentration of aluminum, in real samples. The comparative sorption of aluminum on a novel HPB chelating resin with other solid-phase extractants including XAD-GBH (Islam et al. 2013b) (synthesized in our laboratory) is also reported, wherever required.
Experimental
Chemicals
All chemicals used were of analytical reagent grade (Merck, Mumbai, India). All metal salts were procured from Merck (Mumbai, India) and were standardized by complexometric titration method (Welcher 1958). Working solutions were prepared on a daily basis through serial dilutions of the stock solution with triply distilled water prior to use. The buffer solutions used were hydrochloric acid–glycine, acetic acid–sodium acetate, sodium biphosphate–citric acid, and ammonia–ammonium chloride (Merck, India) for the pH 2.0–3.6, 4.0–5.6, 7.0–7.8, and 8.0–10.0, respectively. Amberlite XAD-16 (particle size 20–60, pore size 100 A°, pore volume 1.82 mL g−1, and surface area 900 m2 g−1) and HPB were procured from Sigma-Aldrich (Steinem, Germany) and Otto Chemicals Pvt. Ltd. (Mumbai, India), respectively. Standard reference materials (SRMs) were obtained from the National Institute of Environmental Studies (NIES) (Ibaraki, Japan) and National Bureau of Standards (NBS), Department of Commerce (Washington, DC, USA).
Apparatus
A flame AAS (M series, Thermo Electron Corporation, Cambridge, UK) equipped with double beam optics and dual Zeeman correction (conditions for Al: a nitrous oxide-acetylene flame; wavelength, 309.3 nm; lamp current, 100 mA; band pass, 0.5 nm; burner height, 11 mm; and fuel flow rate, 4.3 L min−1) was used for determining metal concentration. For batch studies, a thermostated mechanical shaker (NSW-133, New Delhi, India) was used. Thermogravimetric analysis (TGA) and differential thermal analysis (DTA) were done on a Shimadzu TGA/DTA simultaneous measuring instrument (DTG-60/60H, Kyoto, Japan). Elemental (CHN) analysis was carried out on an organic elemental analyzer (Flash EA 1112, Thermo Fischer Scientific). Energy dispersive X-ray analysis (EDAX) was done on a Jeol JSM 6510LV (Japan). A column (1 × 10 cm), for dynamic studies, was obtained from J-SIL Scientific Industries (Agra, India).
Synthesis of chelating resin XAD-HPB
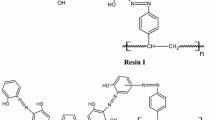
Five grams of oven-dried AXAD-16 was pretreated with an ethanol–hydrochloric acid–water (2:1:1) solution overnight and then rinsed with triply distilled water until the supernatant liquid became neutral to assure the complete absence of impurities. The modification of the pretreated resin was done by initial nitration and then subsequent amination (by refluxing for 12 h in a reducing mixture containing 40 g of SnCl2, 45 mL of concentrated HCl, and 50 mL of ethanol). The amino compound obtained was washed thoroughly with 2 mol L−1 NaOH and then with 4 mol L−1 HCl in order to remove the excess SnCl2. Diazotization of the product was done according to the recommended procedure (Saxena and Singh 1997). After amination, the subsequent steps were carried out at a temperature of 0 to 5 (±0.2) °C in order to prevent the degradation of the intermediates. The diazotized product was rapidly filtered off, washed with cold distilled water until neutral, and then subjected to a coupling reaction by treating it with a solution of HPB (2.5 g) in 10 % NaOH mixed with ethanol at a temperature as low as 0 to 5 (±0.2) °C over a period of 24 h. The reddish brown resin was filtered off and thoroughly washed with 2 mol L−1 HCl and distilled water until pH 7. Finally, the resin was dried at 50 (±0.2) °C and kept in a desiccator for further use. The synthetic scheme is shown in Fig. 1.

Immobilization of HPB ligand on Amberlite XAD-16
Characterization of XAD-HPB
The resin was characterized on the basis of elemental, IR and EDAX analysis, thermal, and chemical studies.
Thermal and chemical stability
The thermal stability of the resin was studied by TGA and DTA analysis. Chemical stability was established by determining the sorption capacity after soaking the resin sets of same amount in 25 mL of acid (1–7 mol L−1 HCl and HNO3) and alkaline solution (1–5 mol L−1 NaOH) for 48 h. The resin was found to withstand several sample injection/elution cycles without discernible loss of performance.
Water regain value and hydrogen ion capacity
The water regain capacity was calculated as mentioned in an earlier work (Islam et al. 2010a). For hydrogen ion capacity, exactly 0.5 g resin was treated with 4.0 mol L−1 HC1 filtered off, washed thoroughly with distilled water, and dried at 100 °C for 6 h. The resin in acidic form was equilibrated with 20 mL of 0.1 mol L−1 NaOH solutions for 6 h and the excess alkali was estimated with 0.1 mol L−1 HCl.
Sample preparation
Fruit juice (3 × 500 mL), peritoneal dialysis fluid (2 × 2,000 mL), and packaged drinking water (3 × 1,000 mL) of different batches were collected from the local market of Aligarh, India. The tap water sample was collected from the university campus. The required volume of each sample after mixing all the contents was then pretreated prior to their application to the SPE procedures. The water samples and dialysis fluid (500 mL each) were filtered through a cellulose membrane filter (Millipore) of 0.45 μm pore size while the fruit juice (500 mL) sample was digested by wet oxidation with 5 mL of conc. HNO3, HClO4, and 2 mL of 30 % H2O2. The residue was dissolved in 5 mL of 0.5 M HNO3 and finally made up to 50 mL with triply distilled water at the working pH. The sample solutions of 2,000 and 55 mg of rice flour-unpolished NIES-10(a) and citrus leaves NBS-1572, respectively, were similarly prepared as reported in earlier work (Islam et al. 2010b).
Recommended procedure
Batch method
Batch experiments were conducted by equilibrating 50.0 mL of metal solution of suitable concentration at constant pH with 0.1 g XAD-HPB in an Erlenmeyer flask stirred for 3 h at 27 ± 0.2 °C. The metal ions were desorbed by shaking with the appropriate eluting agents and measured by FAAS.
Column method
A total of 0.3 g chelating resin was filled into a glass column (10 cm × 1.0 cm) fitted with porous disk. Before use, the column was treated with 10 mL of 2.0 mol L−1 HNO3 solutions and washed with double distilled water until the resin was free from acid. Then, the column was preconditioned to the desired pH with buffer solution. The sample solution was permitted to flow through the column under gravity at set optimum conditions. The column was then rinsed thoroughly by triply distilled water. The metal ions were stripped off from the resin bed by using a set volume of eluent and determined by FAAS.
Result and discussion
Characterization of XAD-HPB
The TGA/DTA curve depicted that thermally the degradation of the functionalized resin commences from 270 °C; above which an exothermic peak (at 380 °C) indicating a weight loss of 51 % was observed (Online Resource 1 and 2). The chemical stability was established up to 5 mol L−1 of mineral acids and alkali. The water regain capacity was calculated to be 21.63 mM g−1 reflecting the high hydrophilic character of the resin which is excellent for column operation (Islam et al. 2010a). The results of elemental analysis of nitrated, aminated (Online Resource 3), and reagent-coupled resin (Online Resource 4) were (C, 67.7; H, 5.9; N, 4.6 %), (C, 58.9; H, 4.6; N, 4.0 %), and (C, 73.2; H, 5.9; N, 4.0 %), respectively. The relative nitrogen percent calculated on comparing theoretical and experimental CHN data resulted in 83, 74, and 45 % efficient conversion for the subsequent steps depicted in Fig. 1, respectively. This inferred that there was an incorporation of one group or reagent molecule per two monomer units. The overall hydrogen ion capacity amounts to 1.7 mM g−1 of resin. This affirmed the incorporation of 0.85 mM reagent g-1 resin, taking into account that 1 mol of reagent is equivalent to 2 mol of replaceable hydrogen ions. To validate the proper synthesis, IR spectra of nitrated (Online Resource 5), aminated (Online Resource 6), and HPB-functionalized AXAD-16 (Online Resource 7) were compared. A peak at 3,380 cm−1 due to –N–H– stretch was observed only in aminated AXAD-16 and was absent in both nitrated and HPB-functionalized AXAD-16. The HPB-functionalized AXAD-16 gave visible peaks at 1,450, 1,708, and 3,417 cm−1 in the IR spectrum, which were ascribed to –N = N–, –C = N–, and phenolic –OH group, respectively, whereas non-functionalized AXAD-16 did not offer any such peaks. Complexation of Al(III) with the synthesized chelating resin was analyzed by EDAX in the range of 0–20 keV. The absence and presence of Al(III) in uncomplexed and complexed XAD-HPB was confirmed by the observations depicted in panels a and b of Fig. 2, respectively.

EDAX spectra showing elemental analysis for the selected area in SEM picture. a EDAX spectra of XAD-HPB. b EDAX spectra of XAD-HPB complexed with aluminum
Optimization studies on the Al(III) ion uptake
Effect of loading solution pH
The effect of pH on the sorption showed that the maximum uptake of Al(III) occurs at pH 9 ± 0.1 (Fig. 3). This is attributed to the involvement of –O− in chelate formation with the Al(III) as a result of the deprotonation of phenol moiety of the reagent (Pearson 1968; Pearson 1963), and to the competition between the hydrogen and aluminum ions on the sorption sites, at low pH (Abdelwahab 2007). For subsequent experiments, pH 9 ± 0.1 was selected as the working pH.

Dependence of sorption capacity on the pH of the solution. (Experimental conditions: sample volume 50 mL, metal ion 100 μg mL−1, resin amount 0.1 g)
Effect of contact time
The contact time of the sorption reaction are one of the important characteristic in defining Al(III) sorption. The results indicated that the solid-phase extraction is very rapid for the highly hydrophilic XAD-HPB: t 1/2 of merely 7 min (Fig. 4). The fast sorption kinetics is an obvious advantage for the preconcentration studies; a time of 10 min (100 % saturation) was then set in all further experiments.

Sorption kinetics for Al(III) complexation on XAD-HPB
Sample flow rate and desorption of Al(III) ions
High sample flow rate is desirable to achieve the highest preconcentration factor in the shortest time. Observations indicated that metal retention on the resin was optimum at a flow rate of 5 mL min−1. In the elution studies, >98 % recovery of the sorbed metals from the resin could be achieved at a flow rate of 2 mL min−1. The percent recovery for aluminum by using 5 mL each of 2 M HCl, 2 M HNO3, and 2 M H2SO4 were found to be 98.9 ± 1, 72 ± 3, and 68 ± 2, respectively. In consequence, the sorption flow rate of 5 mL min−1, the elution flow rate of 2 mL min−1, and 5.0 mL of 2 M HCl as eluent were set in all further dynamic operations.
Physical studies
Langmuir and Freundlich sorption isotherm equilibrium models were used for the analysis of the present sorption system (Langmuir 1918). The Langmuir sorption isotherm model, to evaluate the monolayer sorption phenomena, is depicted in the equation.
where q e is the analyte uptake per unit weight of sorbent (mg g−1), C e is the concentration of analyte in aqueous phase at equilibrium (mg L−1), and q m and K b are Langmuir constants related to the sorption capacity and sorption energy, respectively. The constants can be evaluated from the intercept and the slope of the linear plots of C e/q e vs. C e (Fig. 5). The experimental data shows a straight line with a good correlation coefficient (r 2 = 0.999) indicating the applicability of the model for the present sorption. The essential characteristics of the Langmuir isotherm can be expressed in terms of either a dimensionless constant separation factor or equilibrium parameter

Langmuir sorption isotherm for Al(III) on XAD-HPB
Where R L is a dimensionless separation factor, C 0 is the initial analyte concentration (mg L−1), and K b is the Langmuir constant (L mg−1). The calculated R L values (0 < R L > 1) indicate the favorable sorption of Al(III) onto XAD-HPB. The linear Freundlich equation is
Where k and n are Freundlich constants indicating the sorption capacity and intensity, respectively. Theoretical linear plots, lnq e vs lnC e with low correlation coefficient (r 2 = 0.884) indicates the non-applicability of Freundlich sorption isotherm (Online Resource 8).
The Scatchard plot analysis is also used to investigate the sorption process and nature of binding sites in solid phase. The Scatchard equation is represented as (Gezici et al. 2007)
where q m and K b are the sorption isotherm parameters related to the slope and intercept of q e/C e vs q e (Fig. 6). The type of the interactions of analyte with sorbent is related to the shape of the Scatchard plot. The presence of a deviation from linearity on a plot based on Scatchard analysis usually points to the presence of more than one type of the binding sites, while the linearity of the Scatchard plot observed here indicated that the binding sites are identical and independent. The coordination mode shown in Fig. 1 was further explored by running MM2 of CS Chem 3D Ultra 7.0.0 software for the minimum energy run (7.9) of aluminum ligand complex (Fig. 7).

Scatchard plot for Al(III) on XAD-HPB

CS Chem 3D ultra model of Al(III) complexation on XAD-HPB
Preconcentration and recovery of metal ions
For the determination of trace elements in large volume of real samples, the analyte should be preconcentrated in smaller volume. Preconcentration improves the analytical detection limit, increases the sensitivity by several orders of magnitude, enhances the accuracy of the results, offers a high degree of selectivity, and facilitates calibration (Mizuike 1983; Pyrzynska and Trojanowicz 1999). Under optimized conditions, the preconcentrating ability of the chelating resin along with lower limit of quantification was investigated by diluting the aluminum ion solution and keeping the total amount of loaded metal at 3 μg. Table 1 reflects the preconcentration ability of XAD-HPB with the highest preconcentration factor of 240 when the final volume was 5 mL, thus indicating the resin’s ability to extract analyte quantitatively even from large sample volumes. The detection limit and limit of quantification evaluated as 3 s and 10s (Long and Winefordner 1983) of the mean blank absorbance for 20 replicate measurements (100 mL each), after preconcentration were 2.8 (absorbance = −0.0017) and 9.2 μg L−1, respectively. The enhanced lower limit of aluminum detection value indicates the high sensitivity of the developed method.
Interference
The preconcentration procedures can be substantially affected by various potential concomitants through precipitate formation, redox reactions, or competing complexation reactions, either of interferent anions with the analyte metal ion or of the metal ions in matrix with the sorbent. The aluminum preconcentration was not significantly affected by the most common matrix anions and even in the presence of alkali and alkaline earth metals and few other investigated metal cations (Table 2). This unique selectivity of XAD-HPB towards Al(III), at the optimized conditions, can be interpreted in terms of the smaller ionic radius and higher charge density of the Al(III). The smaller radius of the Al(III) permits suitable coordination geometry for the chelating resin and the larger charge density allows strong coordination ability between XAD-HPB and Al(III).
Analytical figures of merit
The bias of the outlined separation/preconcentration method was estimated by the analysis of trace amount of Al(III) present as major and minor component in studied SRMs. The mean concentration values for Al(III) obtained by the proposed method (Table 3) were statistically insignificant from the certified values indicating absence of systematic method errors. The method also had good precision for the analysis of trace Al(III) in sample solutions, as the coefficient of variation for five replicate measurements of 5 μg of Al in 100 mL was <5 %. The calibration curve with the regression equation and correlation coefficient (r 2) for Al(III) determination, after SPE, obtained by the method of least squares, was A = 0.0043C + 0.0001 (r 2 > 0.99), where A is the absorbance and C is the metal ion concentration (mg L−1). The linearity of the calibration curve is apparent from the correlation coefficient (r 2) which lies well above 0.99.
Application of the method
The preconcentration factor of the presented Al(III) separation/preconcentration is superior in comparison to various matrices shown in Table 5. The samples were analyzed at optimized conditions followed by subsequent determination of eluent in FAAS. To rule out the presence of constant errors, by varying the sample size, the proposed method was applied to the determination of Al(III) in the pretreated sample solutions spiked with known amount of Al(III) in 500 mL of sample solution. Table 4 attested the applicability of the procedure for aluminum determination with good recovery (>95 %).
Conclusion
In this article, HPB-functionalized AXAD-16 chelating resin was synthesized by a simple procedure and was employed as a selective solid-phase extraction material for separation/preconcentration of aluminum in real samples prior to its determination by flame atomic absorption spectrophotometer (Table 5). A brief description of characteristics of the synthesized chelating resin followed by description of application in the selected pretreated real samples is reported. The prepared sorbent offered a fast kinetics for the sorption and desorption of aluminum. The resin could be recycled many times (75 cycles) without affecting its sorption capacity of 21.58 mg g−1. The elution was easily achieved using 5.0 mL of 2 M HCl. Under the optimized conditions, the presence of the major matrix anions and alkali and alkaline earth metals ions did not interfere with the analysis even when interferent cations (Co2+, Mn2+, Ni2+, Cr3+, Pb2+, Cu2+, and Zn2+ ) are 25 times the analyte concentration and, therefore, it was concluded that this analysis could be considered as aluminum selective at optimized condition. The detection limit of 2.8 μg L−1 with the relative standard deviation of less than 5 % (C = 0.05 μg mL−1, n = 5) for aluminum was obtained. To validate the proposed method, certified reference materials were analyzed and the determined values were in a good agreement with the certified values. The developed method offers rapidity, selectivity, sensitivity with high preconcentration factor, and good analyte recovery (>95 %).
References
Abdelwahab, O. (2007). Kinetic and isotherm studies of copper(II) removal from wastewater using various adsorbents. Egyptian Journal of Aquatic Research, 33, 125–143.
Bergmann, A., & Hardt, K. (1979). Analysis of dissolved Cr3+ and Cr6+ in water by APDC-MIBK extraction and atomic absorption spectrometry. Fresenius' Zeitschrift für Analytische Chemie, 297, 381–383.
Boudenne, J., Boussetta, S., Brach-Papa, C., Branger, C., Margaillan, A., & Theraulaz, F. (2002). Modification of poly(styrene-divinylbenzene) resin by grafting on an aluminium selective ligand. Polymer International, 51, 1050–1057.
Carasek, E. (2000). A low-cost flame atomic absorption spectrometry method for determination of trace metals in aqueous samples. Talanta, 51, 173–178.
Chen, J., Huang, C., Hu, B., & Jiang, Z. (2004). Speciation of aluminum in drink samples by 8-hydroxyquinoline loaded silylanization silica gel microcolumn separation with off-line ICP-MS detection. Journal of Agricultural and Food Chemistry, 52, 6843–6847.
Dutta, S., & Das, A. K. (2008). Separation of selected 4f and 5f metals by solid phase extraction: a review. Journal of the Indian Chemical Society, 85, 9–21.
Ejaz, M., Zuha, S., Dil, W., Akhtar, A., & Chaudhri, S. A. (1981). Extraction and preconcentration of copper from water, soils, lubricating oils and plant materials and its subsequent determination by atomic absorption spectrophotometry. Talanta, 28, 441–446.
Exley, C., Merce, A. L. R., Felcman, J., & Recio, M. A. L. (2009). Molecular and supramolecular bioinorganic chemistry. Applications in medical sciences (p. 45). New York: Nova Science Publishers Inc.
Erdemoglu, S. B., Pyrzyniska, K., & Gucer, S. (2000). Speciation of aluminum in tea infusion by ion-exchange resins and flame AAS detection. Analytica Chimica Acta, 411, 81–89.
Gezici, O., Kara, H., Yanik, S., Ayyildiz, H. F., & Kucukkolbasi, S. (2007). Investigating sorption characteristics of copper ions onto insolubilized humic acid by using a continuously monitored solid phase extraction technique. Colloids Surfaces A, 298, 129–138.
Horng, C. J., & Lin, S. R. (1997). Determination of urinary trace elements (As, Hg, Zn, Pb, Se) in patients with Blackfoot disease. Talanta, 45, 75–83.
Hoveyda, H. R., Rettig, S. J., & Orvig, C. (1993). Coordination chemistry of 2-(2-hydroxyphenyl)-2-benzoxazole with gallium(III) and aluminum(III): two uncommon group 13 metal environments stabilized by a biologically relevant binding group. Inorganic Chemistry, 32, 4909–4913.
Henary, M. M., & Fahrni, C. J. (2002). Excited state intramolecular proton transfer and metal ion complexation of 2-(2-hydroxyphenyl) benzazoles in aqueous solution. Journal of Physical Chemistry A, 106, 5210–5220.
Islam, A., Ahmad, A., & Laskar, M. A. (2011a). A newly developed salicylanilide functionalized amberlite XAD-16 chelating resin for use in preconcentration and determination of trace metal ions from environmental and biological samples. Analytical Methods, 3, 2041–2048.
Islam, A., Ahmad, A., & Laskar, M. A. (2012a). Preparation, characterization of a novel chelating resin functionalized with o-hydroxybenzamide and its application for preconcentration of trace metal ions. Clean - Soil, Air, Water, 40, 54–65.
Islam, A., Ahmad, A., & Laskar, M. A. (2012b). Characterization of a chelating resin functionalized via azo spacer and its analytical applicability for the determination of trace metal ions in real matrices. Journal of Applied Polymer Science, 123, 3448–3458.
Islam, A., Laskar, M. A., & Ahmad, A. (2011b). The efficiency of amberlite XAD-4 resin loaded with 1-(2-pyridylazo)-2-naphthol in preconcentration and separation of some toxic metal ions by flame atomic absorption spectrometry. Environmental Monitoring and Assessment, 175, 201–212.
Islam, A., Laskar, M. A., & Ahmad, A. (2013a). Preconcentration of metal ions through chelation on a synthesized resin containing O, O donor atoms for quantitative analysis of environmental and biological samples. Environmental Monitoring and Assessment, 185, 2691–2704.
Islam, A., Laskar, M. A., & Ahmad, A. (2010a). Characterization and application of 1-(2-pyridylazo)-2-naphthol functionalized amberlite XAD-4 for preconcentration of trace metal ions in real matrices. Journal of Chemical and Engineering Data, 55, 5553–5561.
Islam, A., Laskar, M. A., & Ahmad, A. (2010b). Characterization of a novel chelating resin of enhanced hydrophilicity and its analytical utility for preconcentration of trace metal ions. Talanta, 81, 1772–1780.
Islam, A., Ahmad, H., Zaidi, N., & Yadav, S. (2013b). Selective separation of aluminum from biological and environmental samples using glyoxal-bis(2-hydroxyanil) functionalized amberlite XAD-16 resin: kinetics and equilibrium studies. Industrial and Engineering Chemistry Research, 52, 5213–5220.
Langmuir, I. (1918). The sorption of gases on plane surfaces of glass, mica and platinum. Journal of the American Chemical Society, 40, 1361–1403.
Long, G. L., & Winefordner, J. D. (1983). Limit of detection: a closer look at the IUPAC definition. Analytical Chemistry, 55, 712A–724A.
Luo, M., & Bi, S. (2003). Solid phase extraction-spectrophotometric determination of dissolved aluminum in soil extracts and ground waters. Journal of Inorganic Biochemistry, 97, 173–178.
Matus, P., & Kubova, J. (2006). Complexation efficiency of differently fixed 8-hydroxyquinoline and salicylic acid ligand groups for labile aluminium species determination in soils-comparison of two methods. Analytica Chimica Acta, 573–574, 474–481.
Mizuike, A. (1983). Enrichment techniques for inorganic trace analysis. Berlin: Springer.
Martin-Esteban, A., Fernandez, P., Perez-Conde, C., Gutierrez, A., & Camara, C. (1995). On-line preconcentration of aluminium with immobilized Chromotrope 2B for the determination by flame atomic absorption spectrometry and inductively coupled plasma mass spectrometry. Analytica Chimica Acta, 304, 121–126.
Mohammad, B., Ure, A. M., & Littlejohn, D. (1992). On-line preconcentration of aluminium with immobilized 8-hydroxyquinoline for determination by atomic absorption spectrometry. Journal of Analytical Atomic Spectrometry, 7, 695–699.
Neal, C., Rowland, P., Neal, M., Jarvie, H. P., Lawlor, A., Sleep, D., & Scholefield, P. (2011). Aluminium in UK rivers: a need for integrated research related to kinetic factors, colloidal transport, carbon and habitat. Journal of Environmental Monitoring, 13, 2153–2162.
Ottaway, J. M., & Pradhan, N. K. (1973). Determination of chromium in steel by atomic absorption spectrometry with an air acetylene flame. Talanta, 20, 927–931.
Paleologos, E. K., Giokas, D. L., & Karayannis, M. I. (2005). Micelle-mediated separation and cloud-point extraction. Trends in Analytical Chemistry, 24, 426–436.
Popova, S. A., Bratinova, S. P., & Ivanova, C. R. (1991). Determination of trace amounts of copper, nickel and zinc in palladium compounds by solvent extraction flame atomic absorption spectrometry. Analyst, 116, 525–531.
Pearson, R. G. (1968). Hard and soft acids and bases, HSAB, Part II. Journal of Chemical Education, 45, 643–648.
Pearson, R. G. (1963). Hard and soft acids and bases. Journal of the American Chemical Society, 84, 3533–3539.
Pyrzynska, K., & Trojanowicz, M. (1999). Functionalized cellulose sorbents for preconcentration of trace metals in environmental analysis. Critical Reviews in Analytical Chemistry, 29, 313–321.
Pesavento, M., Biesuz, R., Alberti, G., & Sturini, M. (2003). Separation of copper(II) and aluminium(III) from fresh waters by solid phase extraction on a complexing resin column. Journal of Separation Science, 26, 381–386.
Qin, W., Obare, S. O., Murphy, C. J., & Angel, S. M. (2001). Specific fluorescence determination of lithium ion based on 2-(2-hydroxyphenyl)benzoxazole. Analyst, 126, 1499–1501.
Resing, J. A., & Measures, C. I. (1994). Fluorometric determination of Al in seawater by flow injection analysis with in-line preconcentration. Analytical Chemistry, 66, 4105–4111.
Silva, E. L., Grauzarolli, E. M., & Carasek, E. (2004). Use of Nb2O5-SiO2 in an automated on-line preconcentration system for determination of copper and cadmium by FAAS. Talanta, 62, 727–733.
Saxena, R., & Singh, A. K. (1997). Pyrocatechol violet immobilized Amberlite XAD-2: synthesis and metal-ion uptake properties suitable for analytical applications. Analytica Chimica Acta, 340, 285–290.
Sombra, L. L., Wuilloud, R. G., Olsina, R. A., Fernandez, L. P., & Martinez, L. D. (2000). On-line preconcentration system for aluminum determination in parenteral solutions using flow injection-inductively coupled plasma atomic emission spectrometry. Journal of Trace and Microprobe Techniques, 18, 431–436.
Tian, Y., Chen, C. Y., Yang, C. C., Young, A. C., Jang, S. H., Chen, W. C., & Jen, A. K. Y. (2008). 2-(2′-Hydroxyphenyl)benzoxazole-containing two-photon-absorbing chromophores as sensors for zinc and hydroxide ions. Chemistry of Materials, 20, 1977–1987.
Welcher, F. J. (1958). The analytical uses of ethylenediaminetetraacetic acid. New York: Van Nostrand Company.
Yuan, D., & Shuttler, I. L. (1995). Flow-injection column preconcentration directly coupled with electrothermal atomization atomic absorption spectrometry for the determination of aluminium. Comparison of column packing materials. Analytica Chimica Acta, 316, 313–322.
Yaman, M. (2001). Simultaneous enrichment of Cd, Pb, Ni and Al and their determination in water by STAT-FAAS. Spectroscopy Letters, 34, 763–768.
Zhang, X. B., Peng, J., He, C. L., Shen, G. L., & Yu, R. Q. (2006). A highly selective fluorescent sensor for Cu2+ based on 2-(2-hydroxyphenyl)benzoxazole in a poly(vinyl chloride) matrix. Analytica Chimica Acta, 567, 189–195.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Online Resource 1
(GIF 867 kb)
Online Resource 2
(GIF 713 kb)
Online Resource 3
(GIF 703 kb)
Online Resource 4
(GIF 675 kb)
Online Resource 5
(JPEG 354 kb)
Online Resource 6
(JPEG 380 kb)
Online Resource 7
(JPEG 355 kb)
Online Resource 8
(GIF 1204 kb)
Rights and permissions
About this article

Cite this article
Islam, A., Zaidi, N., Ahmad, H. et al. Synthesis, characterization, and systematic studies of a novel aluminum selective chelating resin. Environ Monit Assess 186, 5843–5853 (2014). https://doi.org/10.1007/s10661-014-3823-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10661-014-3823-5