
Abstract
Bacterial canker caused by Clavibacter michiganensis subspecies michiganensis (Cmm) has been a threat to tomato production in Uruguay for several years. In this study, 39 Cmm strains were collected from several outbreaks and production areas in Uruguay, and were identified by molecular assays and pathogenicity tests on a susceptible cultivar of tomato. In addition, a TaqMan assay targeting a putative two-component system sensor kinase gene demonstrated good specificity with all strains tested and gave no false negative results. The first epidemiological study of Cmm in this country was carried out in order to elucidate the origin of outbreaks and sources of infection and dissemination of the pathogen. Strains from Uruguay showed high genetic diversity based on a Multi Locus Sequence Typing analysis of five housekeeping genes. This approach revealed 36 sequence types (STs) within a worldwide collection of 108 Cmm strains. Ten STs correspond to strains solely isolated in Uruguay, including eight novel STs for the subspecies michiganensis. This high diversity reflects the introduction of new strains from different origins that most probably results from seed importation. This study provides relevant information about the distribution and origin of strains causing bacterial canker in Uruguay and will pave the way for the establishment of preventive measures to control the disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Bacterial canker caused by Clavibacter michiganensis subsp. michiganensis (Cmm) is the most important bacterial disease of tomato (Solanum lycopersicum) constituting a worldwide threat to the tomato production (Gleason et al. 1993). Since its first description in Michigan, USA in 1909 (Smith 1910), this disease has been characterized by sporadic occurrence causing severe outbreaks in many countries (Strider 1969). The disease is widely distributed and has been reported in the five continents (EFSA 2014). Recently, outbreaks were reported mostly from Europe and Asia, but also from Latin America and Africa (De León et al. 2011). In Uruguay, tomato bacterial canker was first reported by Lasa et al. (1981) and since then it has emerged sporadically in several fields and greenhouses in the country, although not being formally reported. In recent years, severe outbreaks of the disease occurred in Uruguay, causing substantial economic losses to growers in the two major tomato production areas in the country (Northern and Southern).
Cmm penetrates a plant through wounds or natural openings, reaches the xylem, and develops a massive systemic infection causing the wilting of the whole plant and its death. Once the disease is established in the crop it is very difficult to control and cultural practices, such as eradication of infected plants and disinfestation of materials, are recommended (De León et al. 2011; EFSA 2014). Cmm can survive on plant debris in soil for 2–3 years (Fatmi and Schaad 2002) being a second source of inoculum. Although sources of resistance to bacterial canker have been reported (Sen et al. 2012; van Heusden et al. 1999), there is no resistant tomato cultivar available nowadays (Sen et al. 2015). Other control methods, such as chemical (De León et al. 2008) and biological treatments (Umesha 2006) have been investigated, but no successful control of the disease in tomato has been found yet. Currently, prevention is the most efficient control of bacterial canker, based on the use of healthy seeds and seedlings (De León et al. 2011; EFSA 2014).
At present, this bacterium is a quarantine organism in the European Union, listed as an A2 quarantine pest (EPPO 2013). In Uruguay, most tomato seeds are imported each year from several companies that have their productions centers in different countries (i.e. USA, France, Thailand, Italy, China) and phytosanitary certificates are only required for quarantine pathogens. However, no sanitary measures are applied for other pathogens of agricultural importance, as is the case for bacterial canker (DGSA-MGAP 2009). Several nurseries import seed lots, producing millions of tomato seedlings that are planted in Uruguayan fields. Thus, information regarding the introduction and subsequent spread of bacterial canker is needed. Generally, it is assumed that most outbreaks are caused by infected seeds or seedlings distributed by local nurseries, but the real origin of the disease outbreaks remains unknown.
Characterizing the population structure and genetic diversity of plant pathogens is required to determine possible inoculum sources and design effective disease management strategies. A number of methods have been developed for the identification and characterization of Cmm (Quesada-Ocampo et al. 2012). Previous studies have shown that among DNA-based typing procedures, Pulse Field Gel Electrophoresis (PFGE) (Kleitman et al. 2008), repetitive sequence based PCR (rep-PCR) (Kawaguchi et al. 2010) and Inter Simple Sequence Repeat (ISSR)-PCR fingerprints (Baysal et al. 2011) are suitable for epidemiological studies of Cmm. However, these techniques are time-consuming and the reproducibility of the results among laboratories is low. Multi Locus Sequence Analysis and Typing (MLSA and MLST) developed by Maiden et al. (1998) has proven to be quick, efficient and reproducible approaches for determination of genetic diversity of Cmm strains (Milijašević-Marčić et al. 2012; Jacques et al. 2012). While MLSA relies on the comparison of partial DNA sequences of each gene or of concatenated sequences among strains, MLST is based on the analysis of the combination of alleles at each locus, defining a sequence type (ST). Thus, MLSA provides a framework for species definition and MLST is used to type strains at an infraspecific level. A few MLST schemes have been developed for several pathogens (Almeida et al. 2010; Maiden et al. 1998; Nunney et al. 2012). This technique allows for the unambiguous characterization of isolates from infectious agents using sequences of internal fragments of several housekeeping genes.
Despite severe outbreaks documented in Uruguay in the last years, characterization and diversity studies of Cmm isolates are lacking. There is no data relating to the origin, dissemination and genetic diversity of Cmm strains in Uruguay. Genetic pathogen mapping is an useful tool to follow the flow of new inoculum sources in a country (Baysal et al. 2011). Therefore, the aims of this study were: i) to generate a local collection of Cmm isolates, ii) to characterize the population of Cmm in Uruguay by MLSA and MLST, iii) to compare its genetic variability with other Cmm strains isolated in other countries, and iv) to determine the main inoculum sources for bacterial canker in Uruguay.
Materials and methods
Bacterial strains and growth conditions
Samples of infected tomato plants were collected from several fields and greenhouses of the two major tomato-growing areas in Uruguay in 2010–2012. Strains were isolated from stems of tomato plants showing disease symptoms in Nutrient Broth Yeast (NBY) agar medium (per liter: nutrient broth 8 g, yeast extract 2 g, potassium phosphate dibasic 2 g, potassium phosphate monobasic 0.5 g, pH 7.2 and agar 15 g) with previous disinfestation of the material with hypochlorite at 1 %. Bacteria were grown on NBY at 28 °C for 48–72 h, and conserved at −80 °C in 10 % glycerol for long-term storage. A total of 39 isolates from Uruguay were used in this study (Table 1). Isolates from 1997 to 2008 and some of 2012 were provided by the Ministerio de Ganadería, Agricultura y Pesca (MGAP), Uruguay.
Pathogenicity tests
All strains were inoculated on tomato seedlings S. lycopersicum cv. Elpida. The stems of 4 week-old plants were inoculated at the first true leaf with a toothpick that had been dipped in a fresh colony. Plants were incubated in a growth chamber at 28 °C and symptoms were recorded during 22 days. Four plants were inoculated per strain and water was used as the mock control.
PCR-based assays for identification of Cmm
Total DNA from the 39 Cmm strains was extracted according to Sambrook and Russell (2001). The quantity of extracted DNA was measured by Nanodrop ND-100 (Nanodrop Technologies) and final DNA concentrations were adjusted to 10 ng μl−1 (by dilution) and stored at −20 °C before use.
For identification of the strains, the pair of primers PSA 4/R targeting the Intergenic Spacer region (Pastrik and Rainey 1999) and the universal Eub 27 F/1492R targeting the 16S rRNA were used (Giovannoni 1991) (Table 2). Amplification of the 16S rRNA gene was performed using 25 μl reaction mixtures containing 1X Taq buffer, 2 mM of MgCl2, 200 μM of each dNTP, 0.4 μM of each primer, 0.4 U of Taq polymerase (Invitrogen), and 20 ng of DNA template. The amplification program included an initial denaturation at 94 °C for 3 min; 30 cycles of denaturation (94 °C, 60 s), annealing (55 °C, 60 s), and extension (72 °C, 60 s); and a final extension at 72 °C for 5 min. The PCR products were checked by electrophoresis in 1.5 % agarose in 1X Tris-Borate-EDTA (TBE) and afterwards stained using ethidium bromide. The 16S rRNA products were sequenced with reverse and forward primers by Macrogen Services (Seoul, Korea), and compared in GenBank by the Blastn tool. The PCRs for the amplification of the ITS (PSA 4/R) were followed according to Pastrik and Rainey (1999) and products were processed as explained above.
PCR reactions for detection of pathogenicity genes: pat1, celA, ppaA, chpC, tomA (Kleitman et al. 2008, Table 2), were carried out by the following conditions: initial denaturation step (94 °C, 5 min); 35 cycles of denaturation (94 °C, 30 s), annealing (58 °C, 30 s), and extension (72 °C, 30 s); and a final extension (72 °C, 5 min). PCRs were performed in a final volume of 20 μl containing 1X GoTaq buffer (Promega), 250 μM of each dNTP, 0.25 μM of each primer, 0.4 U of GoTaq polymerase, and 20 ng of DNA template. The PCR products were checked as explained above.
Identification of Cmm strains isolated in Uruguay was done by qPCR using two different sets of primers and probe. The first one targets the putative two-component system sensor kinase (Ptssk) (Berendsen et al. 2011), and is recommended by the International Seed Federation (ISF) (“Method for the Detection of Clavibacter michiganensis subsp. michiganensis on Tomato seed” available online at www.worldseed.org). The reaction was carried out in a final volume of 10 μl containing 2X Master mix (Quanti-Tec, Qiagen), 0.12 μM of each primer and the probe, and 20 ng of DNA template. The run was performed in a Rotor-Gene 6000 real-time rotary analyzer (Corbett Research) with 5 min incubation at 95 °C followed by 40 cycles of 15 s at 95 °C and 30 s at 60 °C. The second test targets the Intergenic Spacer Region (ITS) and reactions were done as described in Luo et al. (2008).
Multi Locus Sequence Analysis (MLSA)
In order to get a better insight into population structure of isolates, further characterization of the collection by MLSA was conducted using five housekeeping genes: atpD (ATP synthase β chain), dnaK (70 kDa heat shock protein), gyrB (DNA gyrase β subunit), ppk (polyphosphate kinase) and recA (recombinase A) (Table 2). PCR amplifications were performed as described in Jacques et al. 2012. PCR products were sequenced in both directions using primers described in Table 2 and Sanger technology by Genoscreen (Lille, France).
Sequences were assembled, edited and aligned using Geneious Pro 4.8.5 software (Biomatters) to obtain high quality sequences. In order to compare the genetic variability of the strains from Uruguay with other Cmm strains isolated in other countries, the corresponding partial sequences of 89 strains from the B-collection of Jacques et al. 2012 were used. This collection includes strains from all subspecies of C. michiganensis: michiganensis (69 strains), insidiosus (3 strains), sepedonicus (3 strains), tessellarius (3 strains) and nebraskensis (4 strains), 6 Clavibacter-like strains and Rathayibacter iranicus as outgroup used to root phylogenetic trees. New multiple alignments for each gene were generated by ClustalW in BioEdit Sequence Alignment Editor 7.0.4.1 software (Hall 1999). Nucleotide alignments were translated to amino acid sequences to gain a codon based alignment (Hall 1999). Sequences were concatenated using Geneious software, following the alphabetic order of the genes and ending in a large sequence of 3009 bp (atpD, dnaK, gyrB, ppk, and recA).
Phylogenies of each loci and concatenated sequences were inferred by maximum likelihood (ML). Best evolution models for each alignment (five loci and the concatenated) were inferred by ModelTest 3.7 in PAUP4 (Swofford 2002) using the hierarchical Likelihood Ratio Tests (hLRT) as well as the Akaike Information Criterion (AIC). Bootstrap analyses were done with 1000 replicates. Trees were generated with PhyML (Guindon et al. 2010). To test if topology of the tree based on the concatenated data set was congruent with the trees produced for each loci, the Shimodaira-Hasegawa test (Shimodaira and Hasegawa 1999) was implemented in DNAML program from PHYLIP package (Felsenstein 2005). Sequence diversity and neutrality estimates and statistics were calculated using the program DnaSP v5 (Rozas et al. 2003).
Multi Locus Sequence Typing (MLST)
MLST was carried out by the allele based method as described by Jacques et al. (2012). Each unique sequence of a locus was assigned an allele number, and each different combination of allele’s numbers defined the Sequence Type (ST) in the concatenated data. For this purpose, sequence data was entered in an MLST database to acquire a ST profile (http://pubmlst.org/analysis/). The relatedness among STs was displayed using the Based Upon Related Sequences Types (eBURST V3) approach (Feil et al. 2004). The program infers patterns of evolutionary descent among isolates using a simple model of clonal expansion and diversification (Pérez-Losada et al. 2013).
Nucleotide sequence accession numbers
The sequences reported here have been deposited in the GenBank database with the following accession numbers: KP789619-KP789657, KP828202-KP828240, KP828241-KP828279, KP828280-KP828318, KP828319-KP828357 for atpD, dnaK, gyrB, ppk, and recA genes, respectively.
Results
Characteristics of Cmm strains from Uruguay
The survey of tomato growing regions in Uruguay during the period 2010–2012 performed in this study and the input of additional isolates from previous outbreaks allow us to generate the first local collection of Cmm strains affecting tomato crops in Uruguay (Table 1). Preliminary identification was performed by comparison of 16S rRNA sequences using the Blast tool in GenBank. All strains showed 99–100 % identity with the Cmm reference strain NCPPB 382 (AM711867) and were pathogenic on tomato, causing typical bacterial canker symptoms. In the pathogenicity test that was performed, the plantlets showed wilt symptoms 14 days post-inoculation (dpi), and the whole plants were completely wilted 21 dpi (Fig. 1).

Disease symptoms in tomato plantlets inoculated with a Clavibacter michiganensis subsp. michiganensis strain isolated in Uruguay: a whole plants showing wilting symptoms 14 dpi and b unilateral wilting of the leaves, a typical bacterial canker symptom
Most strains generated products of the expected size by amplification with specific primers targeting the pathogenicity genes pat1, celA, ppaA, tomA and chpC; and with the ITS primers PSA4-R. The only exception was strain MAI1029, for which celA gene was not amplified. Several methods used regularly for the identification of Cmm led to false negatives or false positives (Sen et al. 2015). In this study we used a Taqman assay based on the Ptssk system, for which previous studies have demonstrated a 100 % specificity within a worldwide collection (Jacques et al. 2012). All strains reacted positively in this qPCR reaction, supporting the good specificity of these primers. An additional Taqman system designed by Luo et al. (2008) was also assayed in this study. However, this method did not reveal good specificity within the probe for several strains, which showed atypical curves in the fluorescence plot (Supplemental material, Figure S1).
Multi Locus Sequence Analysis and Typing
The phylogenetic tree based on the data set of concatenated sequences presented a phylogenetic history strongly supported by high bootstrap values (98), and clearly differentiated Cmm from the four other subspecies and from the saprophytes within C. michiganensis (Fig. 2a). Similar results were found using the Neighbor Joining algorithm (data not shown). It is important to note that strains from Uruguay were grouped in a cluster with Cmm strains originating from various origins. These results provide additional confirmation of the identity of the strains. Phylogenetic trees built for each of the five loci with ML algorithm are shown in Figure S2. The Shimodeira-Hasegawa test showed that all trees were significantly incongruent with each other (P values lower than 0.05) but were not significantly different from the tree based on the concatenated data set (Table 3). Hence, the tree based on the data set of concatenated sequences did not contradict the information brought by each gene.

Maximum likelihood (ML) tree based on concatenation of partial sequences of atpD, dnaK, gyrB, ppk and recA genes for a Clavibacter michiganensis strains isolated in Uruguay (called MAI and marked with a grey point) and from a worldwide collection (Jacques et al. 2012) and b the subspecies michiganensis. Only bootstrap values higher than 50 % are displayed at each node in red. Strains from different locations in Uruguay are distinguished by different colors (Salto green, Artigas blue, Montevideo red, Canelones yellow and San José pink)
The set of 108 Cmm isolates (the 69 strains from Jacques et al. (2012) and the 39 strains isolated in Uruguay) was selected to perform further population structure analysis within the subspecies michiganensis. The statistics, diversity estimates and neutrality tests for each alignment (for each loci and the concatenated data set) are presented in Table 4 and the corresponding subtree in Fig. 2b. The genetic diversity was low, as indicated by the level of polymorphism in the analyzed loci (55 polymorphic sites in 3009 total sites). The percentage of polymorphic sites was between 1 and 2 % for atpD, dnaK, recA and gyrB genes. The ppk locus showed the highest variability (4.2 %), having 24 polymorphic sites in 564 total sites. Between eight and 11 haplotypes were detected for each gene, and a total of 36 haplotypes for the data set of concatenated sequences. Tajima’s D and further neutrality tests indicated that all genes presented no significant deviations from neutrality (Table 4). We adopted an approach called eBURST to represent the relatedness between the 108 Cmm strains (Fig. 3). This approach divides an MLST data set into groups of related isolates and clonal complexes, predicts the founding genotype of each clonal complex (CC), and computes the bootstrap support for the assignment (Feil et al. 2004). In this study, 4 CCs (single-locus variants) were identified, linking 2, 5 or 6 STs (Fig. 3). Altogether, these 4 CCs clustered 54 strains representing 50 % of the Cmm strains. In these CCs, strains isolated in different countries, even different continents, over a long period of time, were grouped. Concerning the analysis of double-locus variants (DLV), 103 strains were linked, representing the 95 % of Cmm strains. All other STs were singletons. Two out of four CCs (A and B) were previously identified by Jacques et al. (2012). In this study we found two additional CCs (C and D) due to the new STs assigned to Uruguayan strains (Fig. 3).

e-Burst diagram for 108 Clavibacter michiganensis subsp. michiganensis strains based on alleles of five housekeeping genes (atpD, dnaK, gyrB, ppk and recA genes). Each point represents a sequence type and their relatedness is represented by thick purple lines (clonal complexes, single-locus variation) and sky-blue lines (double-locus variation). Sequence types representing strains from Uruguay are marked with a green point
Diversity among Uruguayan isolates
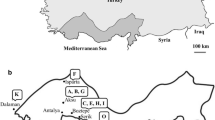
MLST could resolve the 39 Uruguayan isolates into 10 STs, showing high diversity within the Cmm population affecting tomato crops in the country (Table 1; Fig. 4). Most STs and the corresponding CCs were not related to the geographical origin of strains or the year of isolation (Table 1, Fig. 3). This suggests that different sources of inoculum were involved in bacterial canker outbreaks in Uruguay. Strains isolated from a same place fell in separated STs and CCs indicating that inoculum sources might have been diverse in these places. This was the case for strains isolated in Salto (in 2011 and 2012) where STs 1 to 6 were assigned. Strains assigned to ST3 were exclusively isolated in Salto (2012), although they were collected in different farms suggesting a common origin of infection or dissemination for these farms. In the other hand, some STs were not restricted to a particular region; they are present all over the country. For i.e strains assigned to ST5 were isolated from Salto, San José, Canelones and Montevideo (Fig. 4).

Geographical distribution of Clavibacter michiganensis subsp. michiganensis in Uruguay as determined by Multilocus Sequence Typing. Numbers represented the assigned sequenced types
Comparing the genetic diversity of Cmm isolates from Uruguay with a worldwide collection
Thirty one out of the 39 strains from Uruguay presented novel haplotypes compared to the 89 strains of the worldwide B-collection of Jacques et al. 2012. The analysis assigned these 31 strains into 8 STs, which were not established previously (Table S1, Fig. 2b). On the other hand, 8 strains isolated in Uruguay were grouped together with strains from various origins. The most noticeable case is strain MAI1022 (Artigas, 1997) that has the same ST (ST 7) than strains from different countries (Algeria, France, The Netherlands, Belgium and USA) (Table S1; Fig. 2b). This result strongly reflects Cmm seed transmission and hence, the global dissemination of the pathogen through seeds. In another case, a group of strains from Uruguay also shares the same ST with strain CFBP 5843 (Brazil, 1994), which suggests the possibility of material exchange between these neighboring countries or maybe the same source of inoculum.
Discussion
Despite the fact that bacterial canker has been a problem in Uruguay since the early 80s, this is the first report on the genetic diversity of Clavibacter michiganensis subsp. michiganensis in the country. The first collection of Cmm strains from Uruguay that affected tomato crops in recent years was generated and characterized. Several identification tests previously designed for Cmm and the presence of pathogenicity determinants were checked, as well as pathogenicity on tomato.
The development of reliable methods to detect Cmm in seeds (or seedlings) is an important issue to consider, as tomato infected seeds act as source of primary inoculum (Gleason et al. 1993). In particular, the Ptssk system (Berendsen et al. 2011) seems to be the most reliable tool for identification of Cmm. In our experiments, some of the strains isolated in Uruguay had a mismatch of one base in the CMM probe designed by Luo et al. (2008) (Supplemental material, Figure S1). This mismatch led to low amplification efficiencies in the qPCR reactions, explaining the atypical curves obtained for these strains.
In this report, we wanted to decipher how diverse are Cmm isolates recovered in Uruguay and the relationships of these strains to previously characterized strains from a worldwide collection. These elements will be useful to identify the origin of outbreaks in Uruguay. MLST has proven to be a powerful tool for documenting the genetic variability of bacterial isolates. Moreover, it provides an unambiguous “barcode” that identifies any given culture to the level of sequence type (Nunney et al. 2012). Using MLST, phylogenetic relationships of large sets of strains can be analyzed with high reproducibility between laboratories, as demonstrated in this study. The use of housekeeping genes has been adopted for bacterial population studies as they are believed to evolve neutrally. These genes are under stabilizing selection and are present in all strains of a relevant genus/species to be analyzed (Quesada-Ocampo et al. 2012). Still, putative pathogenicity genes are much more conserved than housekeeping genes having a few or no polymorphic sites in the sequenced regions of genes (Tancos et al. 2015). A previous study showed that gyrB and recA genes provided robust phylogenies to identify infraspecies sequence variation among the Clavibacter genus (Jacques et al. 2012). The present study is focused on the diversity among the subspecies michiganensis, so the more polymorphic a gene is, the more informative it is for our purpose. Hence, five housekeeping genes were chosen to compare our collection with strains from diverse origins. In this case, ppk was the most polymorphic gene of this MLST scheme, with 4.3 % of variable sites (Table 4).
Based on this MLST scheme, 108 strains belonging to the subspecies michiganensis were divided in 36 STs. In this context, 10 STs were identified for the 39 isolates from Uruguay, indicating that the Cmm population used in this study has high genetic diversity. Moreover, 8 of these were novel STs expanding the knowledge about the diversity among the subspecies michiganensis. Cmm population structure analyses made in other countries have shown diverse results depending on the amount of strains analyzed and the method used to type them. First, high homogeneity was found in Canary Islands where only one cluster was found for 54 strains using RAPD, BOX-PCR and AFLP; suggesting that Cmm was introduced to Canary Islands from a unique origin (De León et al. 2009). Then, high diversity was found in Japan dividing 43 strains into 22 groups by ISSR-PCR and rep-PCR (Kawaguchi and Tanina 2014). Furthermore, an MLSA scheme made in New York clusters 51 isolates into 21 haplotypes, by using five housekeeping genes (kdpA, sdhA, dnaA, ligA and gyrB) and three putative pathogenicity genes (celA, tomA and nagA) (Tancos et al. 2015). These results as well as the ones obtained in this study, indicate multiple origins of infection for the development of bacterial canker in those geographical places.
A further advantage of MLSA/MLST is that new isolates can be easily grouped into an existing framework of strains. In Uruguay, further isolations remain to be done, and hence the approach reported in this work acquires great relevance to continue determining the origin of new outbreaks of bacterial canker in the country. Interest in knowing the origin of outbreaks has emerged due to real-life problems experienced by growers in the two major production areas, as economic losses caused by this bacterium increase in each growing season. Having the opportunity to use MLSA to identify strains could help growers by demonstrating if the same or a different strain of this pathogen is established on their fields/greenhouses. Depending on the results, growers can know whether the outbreak was caused by improper sanitation measures or by an introduction of the pathogen through infected seeds.
It is well known that contaminated seeds are the primary source of inoculum for most Cmm outbreaks (De León et al. 2011). In Uruguay, many tomato varieties from different countries are introduced each year as a consequence of importation of seeds. From 2010 to 2012 many lots came from Thailand, Peru, Italy, China, Argentina, Denmark, USA, Japan or the Netherlands. We observed high genetic diversity among the strains from Uruguay (Table 1) and hence, introduction and subsequent transmission of the pathogen by seeds is suggested. Probably, the pathogen was transmitted from several of those seed lots and multiplied in nurseries, providing latently infected tomato plants to growers.
There is another possibility regarding the sources of inoculum, the existence of dominant strains surviving in soil or plant debris, being recovered in consecutive years in the same location. This fact was reported in Israel, where two genotypes where repeatedly isolated for several years in the Besor region (Kleitman et al. 2008). We did not find this type of results in our study for the two places where isolations were made in consecutive years (Montevideo and Salto). Despite, isolates from Montevideo collected in 2005 and 2010 were grouped together; the exact location of these strains is not available. In consequence, we cannot conclude for this particular situation. Surprisingly, strains collected in one location (Tropezón) in Salto two following seasons (2011 and 2012) belonged to different STs each year (Table 1). This finding suggests that each year new inoculum is brought to the greenhouse.
Another relevant issue is the geographical distribution of haplotypes over the country. As observed in Fig. 4, most STs were found in the two main tomato growing areas of the country (Southern and Northern). In particular, ST5 was isolated in all locations sampled except Artigas. In addition, some STs seem to be exclusive of one specific location, for instance ST3 was only found in Salto. It is important to notice that both regions have different crop cycles because temperatures in the North are always higher than in the South. In the latter area, tomato is a season crop, so it is harvested from summer to beginning of autumn. The northern region is the major producer of counter season crops to sell during winter and spring, and most part is produced in greenhouses. Hence, it remains to be elucidated if some STs are better adapted to a particular region given by its climate conditions, crop systems, etc.
Phylogenetic trees are not suitable to represent recent evolutionary events, as they attempt to reconstruct relationships in the absence of a realistic model of the way in which bacterial clones emerge and diversify to form clonal complexes (Feil et al. 2004). For this reason, we have used the e-Burst algorithm, which revealed some differences with the one showed in the previous report (Jacques et al. 2012). The discovery of ST3 in this work, allowed the identification of the CC “C”, with ST3 being the ancestral haplotype from which other strains arise. The other strains involved in this CC (those assigned to STs 7, 34, 38, 29 and 48) were not linked altogether in the previous report (Jacques et al. 2012). Furthermore, CC “D” corresponding solely to strains from Uruguay was also identified in this work. Strains forming clonal complexes from different geographical regions and periods of time, illustrates the efficacy of seed transmission and powerful survival strategies.
This study also confirms the great potential of MLST to reveal the origin and spread of bacterial canker. A unique MLST scheme should be adopted for Cmm strains in order to compare strains from worldwide origins in future studies. In conclusion, the main source of bacterial canker infections in Uruguay consisted in infected seeds as a result of importation from different countries. We suggest that a traceability system should be applied to seeds in order to search for origin of outbreaks and follow the spread of possible sources of contamination.
Abbreviations
- Cmm:
-
Clavibacter michiganensis subsp. michiganensis
- ST:
-
Sequence Type
- CC:
-
Clonal complex
References
Almeida, N. F., Yan, S., Cai, R., Clarke, C. R., Morris, C. E., Schaad, N. W., et al. (2010). PAMDB, a multilocus sequence typing and analysis database and website for plant-associated microbes. Phytopathology, 100(3), 208–215.
Baysal, Ö., Mercati, F., İkten, H., Yıldız, R. Ç., Carimi, F., Aysan, Y., & Teixeira da Silva, J. (2011). Clavibacter michiganensis subsp. michiganesis: tracking strains using their genetic differentiations by ISSR markers in Southern Turkey. Physiological and Molecular Plant Pathology, 75(3), 113–119.
Berendsen, S. M. H., Koenraadt, H., Woudt, B., & Oosterhof, J. (2011). The development of a specific Real-Time TaqMan for the detection of Clavibacter michiganensis subsp . michiganensis. Presented at the American Phytopathological Society - International Plant Protection Convention Meeting, Honolulu, Hawaii, 6–10 August 2011.
De León, L., Siverio, F., López, M. M., & Rodríguez, A. (2008). Comparative efficiency of chemical compounds for in vitro and in vivo activity against Clavibacter michiganensis subsp. michiganensis, the causal agent of tomato bacterial canker. Crop Protection, 27(9), 1277–1283.
De León, L., Rodríguez, A., Llop, P., López, M. M., & Siverio, F. (2009). Comparative study of genetic diversity of Clavibacter michiganensis subsp. michiganensis isolates from the Canary Islands by RAPD-PCR, BOX-PCR and AFLP. Plant Pathology, 58(5), 862–871.
De León, L., Siverio, F., López, M., & Rodríguez, A. (2011). Clavibacter michiganensis subsp. michiganensis, a seedborne tomato pathogen: healthy seeds are still the goal. Plant Disease, 95(11), 1328–1338.
DGSA-MGAP (Dirección General de Servicios Agrícolas, Ministerio de Ganadería, Agricultura y Pesca) (2009). Sub-estándar fitosanitario Mercosur. Sección III - Medidas Fitosanitarias. Requisitos fitosanitarios para Lycopersicon esculentum (tomate) según País de Destino y Orígen, para los Estados Partes. http://www.mgap.gub.uy/dgssaa/Normativa/Archivos/INTERN_ResGMC/ResDGSA16tomate.pdf. Accessed 26 Feb 2015.
Dreier, J., Bermpohl, A., & Eichenlaub, R. (1995). Southern hybridization and PCR for specific detection of Clavibacter michiganensis subsp. michiganensis. Phytopathology, 85, 462–468.
EFSA PLH Panel (EFSA Panel on Plant Health). (2014). Scientific Opinion on the pest categorisation of Clavibacter michiganensis subsp. michiganensis (Smith) Davis et al. EFSA Journal. doi:10.2903/j.efsa.2014.3721.
EPPO (European and Mediterranean Plant Protection Organization). (2013). PM 7/42 (2) Clavibacter michiganensis subsp. michiganensis. EPPO Bulletin, 43(1), 46–67.
Fatmi, M., & Schaad, N. W. (2002). Survival of Clavibacter michiganensis ssp. michiganensis in infected tomato stems under natural field conditions in California, Ohio and Morocco. Plant Pathology, 51(2), 149–154.
Feil, E. J., Li, B. C., Aanensen, D. M., Hanage, W. P., & Spratt, B. G. (2004). eBURST: Inferring Patterns of Evolutionary Descent among Clusters of Related Bacterial Genotypes from Multilocus Sequence Typing Data. Journal of Bacteriology, 186(5), 1518–1530.
Felsenstein, J. (2005). PHYLIP: Phylogeny inference package (Version 3.63). Seattle: Department of Genetics, University of Washington.
Fu, Y. X., & Li, W. H. (1993). Statistical tests of neutrality of mutations. Genetics, 133, 693–709.
Giovannoni, S. (1991). The polymerase chain reaction. In E. Stackebrandt & M. Goodfellow (Eds.), Nucleic acid techniques in bacterial systematics (pp. 177–204). Chichester: Wiley.
Gleason, M., Gitaitis, R., & Ricker, M. (1993). Recent progress in understanding and controlling bacterial canker of tomato in eastern North America. Plant Disease, 77(11), 1069–1076.
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., & Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Systematic Biology, 59(3), 307–321.
Hall, T. (1999). BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series.
Jacques, M.-A., Durand, K., Orgeur, G., Balidas, S., Fricot, C., Bonneau, S., et al. (2012). Phylogenetic analysis and polyphasic characterization of Clavibacter michiganensis strains isolated from tomato seeds reveal that nonpathogenic strains are distinct from C. michiganensis subsp. michiganensis. Applied and Environmental Microbiology, 78(23), 8388–8402.
Kawaguchi, A., & Tanina, K. (2014). Genetic groups of Clavibacter michiganensis subsp. michiganensis identified by DNA fingerprinting and the effects of inoculation methods on disease development. European Journal of Plant Pathology, 140(3), 399–406.
Kawaguchi, A., Tanina, K., & Inoue, K. (2010). Molecular typing and spread of Clavibacter michiganensis subsp. michiganensis in greenhouses in Japan. Plant Pathology, 59, 76–83.
Kleitman, F., Barash, I., Burger, A., Iraki, N., Falah, Y., Sessa, G., et al. (2008). Characterization of a Clavibacter michiganensis subsp. michiganensis population in Israel. European Journal of Plant Pathology, 121(4), 463–475.
Lasa, C. I., Abiko, K., Tezuka, N., Inaba, T., & García, S. (1981). Algunas enfermedades que afectan actualmente los cultivos de hortalizas en Uruguay. Investigaciones Agronómicas. Uruguay, Año, 2(Nro 1), 97–100.
Luo, L. X., Walters, C., Bolkan, H., Liu, X. L., & Li, J. Q. (2008). Quantification of viable cells of Clavibacter michiganensis subsp. michiganensis using a DNA binding dye and a real-time PCR assay. Plant Pathology, 57(2), 332–337.
Maiden, M., Bygraves, J., Feil, E., Morelli, G., Rusell, J., Urwin, R., et al. (1998). Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proceedings of the National Academy of Sciences of the United States of America, 95(6), 3140–3145.
Milijašević-Marčić, S., Gartemann, K.-H., Frohwitter, J., Eichenlaub, R., Todorović, B., Rekanović, E., & Potočnik, I. (2012). Characterization of Clavibacter michiganensis subsp. michiganensis strains from recent outbreaks of bacterial wilt and canker in Serbia. European Journal of Plant Pathology, 134(4), 697–711.
Nunney, L., Elfekih, S., & Stouthamer, R. (2012). The importance of multilocus sequence typing: cautionary tales from the bacterium Xylella fastidiosa. Phytopathology, 102, 456–460.
Pastrik, K. H., & Rainey, F. A. (1999). Identification and differenciation of Clavibacter michiganensis subspecies by Polymerase chain reaction- based techniques. Journal of Phytopathology, 147, 687–693.
Pérez-Losada, M., Cabezas, P., Castro-Nallar, E., & Crandall, K. A. (2013). Pathogen typing in the genomics era: MLST and the future of molecular epidemiology. Infection, Genetics and Evolution, 16, 38–53.
Quesada-Ocampo, L. M., Landers, N. A., Lebeis, A. C., Fulbright, D. W., & Hausbeck, M. K. (2012). Genetic structure of Clavibacter michiganensis subsp. michiganensis populations in Michigan commercial tomato fields. Plant Disease, 96(6), 788–796.
Rozas, J., Sánchez-DelBarrio, J., Messeguer, X., & Rozas, R. (2003). DnaSP: DNA polymorphism analysis by the coalescent and other methods. Bioinformatics, 19, 2496–2497.
Sambrook, J., & Russell, D. W. (2001). Molecular Cloning a Laboratory Manual (3rd ed.). New York: Cold Spring Harbor.
Sen, Y., Feng, Z., Vandenbroucke, H., Wolf, J., Visser, R. G. F., & Heusden, A. W. (2012). Screening for new sources of resistance to Clavibacter michiganensis subsp. michiganensis (Cmm) in tomato. Euphytica, 190(2), 309–317.
Sen, Y., van der Wolf, J., Visser, R. G. F., & van Heusden, A. W. (2015). Bacterial canker of tomato: current knowledge of detection, management, resistance and interactions. Plant Disease, 99(1), 4–13.
Shimodaira, H., & Hasegawa, M. (1999). Multiple comparisions of log likelihoods with applications to phylogenetic inference. Molecular Biology and Evolution, 16, 1114–1116.
Smith, E. F. (1910). A new tomato disease of economic importance. Science, 31, 794–796.
Strider, D. L. (1969). Bacterial canker of tomato caused by Corynebacterium michiganense. A literature review and bibliography. North Carolina Agricultural Experiment Station Technical Bulletin 193.
Swofford, J. L. (2002). PAUP. Phylogenetic analysis using Parsimony.
Tajima, F. (1989). Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics, 123, 585–595.
Tancos, M. A., Lange, H. W., & Smart, C. D. (2015). Characterizing the genetic diversity of the Clavibacter michiganensis subsp. michiganensis population in New York. Phytopathology, 105(2), 169–179.
Umesha, S. (2006). Occurrence of bacterial canker in tomato fields of Karnataka and effect of biological seed treatment on disease incidence. Crop Protection, 25(4), 375–381.
Van Heusden, A. W., Koornneef, M., Voorrips, R. E., Brüggemann, W., Pet, G., Vrielink-van Ginkel, R., et al. (1999). Three QTLs from Lycopersicon peruvianum confer a high level of resistance to Clavibacter michiganensis ssp. michiganensis. Theoretical and Applied Genetics, 99(6), 1068–1074.
Acknowledgments
This work was supported by CSIC I + D Grupos N° 652 and MSc. scholarship provided to V. Croce by the National Research Council in Uruguay (ANII).
We thank Enrique Verdier from DGSA-MGAP and Elisa Silvera and María José Montelongo from Agronomy Faculty for providing Cmm strains isolated in Uruguay; Ana Arruabarrena for helping in the isolations of strains from Salto; Sophie Bonneau for her useful advices to build the trees; Eliana Richard for technical support in figure editing; Federico Boschi and Diego Maeso for useful discussions; and Maria Inés Lapaz and Analía Sanabria for critically reviewing the manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Figure S1
qPCR amplification with primers designed by Luo et al. (2008) showing the results of six strains as an example. A. Fluorescence vs. cycles graph showing the inefficiency of the CMM probe by atypical curves (strains MAI 1001, MAI 1003 and MAI 1027). Strains showing typical curves (MAI1031, MAI1029 and MAI1039) are displayed in this graph in order to compare both kinds of curves. B. The six products sequences of the strains showed in (a) aligned and mapped with the probe shows a mismatch in the sequences of strains showing atypical curves. (GIF 80 kb)
Figure S2
Maximum likelihood trees based on partial sequences of genes atpD, dnaK, gyrB, ppk and recA. Bootstrap values higher than 50 are displayed at each node in red. (PDF 423 kb)
Supplemental Table S1
(DOC 167 kb)
Rights and permissions
About this article

Cite this article
Croce, V., Pianzzola, M.J., Durand, K. et al. Multilocus Sequence Typing reveals high variability among Clavibacter michiganensis subsp. michiganensis strains affecting tomato crops in Uruguay. Eur J Plant Pathol 144, 1–13 (2016). https://doi.org/10.1007/s10658-015-0738-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10658-015-0738-0