
Abstract
Botrytis spp. associated with neck rot disease were isolated from New Zealand onions. The fungi were identified using molecular sequences of the ribosomal internal transcribed spacer (ITS) and intergenic spacer (IGS) regions, and the glyceraldehyde-3-phosphate dehydrogenase (G3PDH) gene. Analyses of the sequences showed that the majority of the isolates gathered in 2005–07 were B. aclada. A new high resolution melting analysis (HRMA) assay was developed that allowed fast and simple discrimination between B. aclada and other Botrytis spp. causing onion neck rot in New Zealand. To further verify these results, Botrytis isolates from New Zealand onions, stored in the International Collection of Microorganisms from Plants (ICMP), were also examined. Only a single isolate from the ICMP collection was B. aclada while two isolates were B. byssoidea, one B. squamosa and another closely related to Botryotinia porri. Identification of the remaining Botrytis isolates was more difficult; while IGS and ITS sequences indicated a close relationship to B. allii or B. byssoidea, a previously unreported intron insertion was observed at the 3′ end of the ribosomal small subunit gene in these isolates. No evidence of heterogeneity was observed in the G3PDH gene sequences, as might have been expected of the allodiploid B. allii, but the G3PDH sequence ruled out B. byssoidea as the identity of these isolates.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Botrytis spp. of fungi cause several important diseases of onion (Allium cepa L.), including leaf blight and neck rot of bulbs (e.g. Lorbeer et al. 2004). Symptoms of neck rot include soft and watery decay, white mycelia between the bulb scales, and masses of black sclerotia on the outer scales. The disease, which is often exacerbated by secondary pathogens (Viljanen-Rollinson et al. 2007), may be latent in the field and only become evident after months of storage. Entry of Botrytis into onion bulbs occurs through several routes, including via onion flowers which provide a direct route to seed transmission (Maude and Presly 1977; Stewart and Franicevic 1994; du Toit et al. 2004). Contamination of onion seed by Botrytis spp. has been widely detected and confirmed as a source of inoculum for neck rot in the subsequent crop (Maude and Presly 1977; Stewart and Franicevic 1994; du Toit et al. 2004). The incidence of seedling infection is generally lower than infestation in the preceding seed stock, with severity and incidence of disease dependent on seasonal environmental variables. Other sources of inoculum are cull piles, infected crop residues and sclerotia that may survive in the soil for several years (Lorbeer et al. 2004).
Onions are affected by at least seven Botrytis spp., of which three—B. aclada, B. allii and B. byssoidea — are exclusively associated with neck rot (Chilvers and du Toit 2006). The taxonomy of these three species has been subject to change, with the closely related B. aclada and B. allii first differentiated based on spore dimensions. The large-spored B. allii was also observed to have twice (32) the haploid chromosome number of both the smaller spored B. aclada and B. byssoidea (Shirane et al. 1989). However, because spore size is somewhat plastic, with overlap between isolates of B. allii and B. aclada, this character is insufficient for accurately distinguishing these species.
Molecular markers are now commonly used to characterise Botrytis isolates. A single nucleotide polymorphism in the ribosomal internal transcribed spacer (ITS) fragment distinguishes B. aclada from other onion-infecting Botrytis spp. (Nielsen et al. 1999). Restriction fragment length digestion (RFLP) of the ITS fragment may be used to identify B. aclada versus B. allii. Universally primed PCR (UP-PCR) patterns also showed differences between B. allii and B. aclada and a clear separation of these isolates from B. byssoidea, B. cinerea and B. squamosa (Nielsen et al. 2001). Nielsen and Yohalem (2001) cloned and sequenced a UP-PCR fragment from B. aclada and then developed a sequence characterised amplified region (SCAR)-RFLP assay that is now frequently used to identify onion-infecting Botrytis spp. (e.g. Chilvers et al. 2004). Both UP-PCR patterns and SCAR-RFLP are concordant with ITS-RFLP (Nielsen et al. 2001).
Sequence variation in UP-PCR products led to the further conclusion that B. allii was derived from a hybridisation event between B. aclada and B. byssoidea (Nielsen and Yohalem 2001; Yohalem et al. 2003). This view was bolstered by Staats et al. (2005) who found hybrid sequences in three B. allii genes, especially the HSP60 gene. Polymorphic sites were also observed in the HSP60 genes from B. aclada isolates, but since these fragments were directly sequenced, no precise measure of polymorphism was obtained for this species.
Despite the availability of these molecular tools, identification of the species causing neck rot disease has infrequently been incorporated into field studies of this disease (Chilvers and du Toit 2006). Here we were interested in designing tools for discriminating Botrytis spp. and for the early detection of disease. The identity of Botrytis associated with New Zealand onions has not been investigated using molecular tools and we sought new DNA sequence information for the development of our assays.
Materials and methods
Fungal isolation
Onion bulbs and seeds were progressively obtained from seed companies, exporters and growers during the 2005–07 growing seasons. Of the 48 isolates from this collection that were examined, 16 were from bulbs and 32 from seed. These were sourced mainly from Canterbury, Marlborough and Pukekohe, as well as four isolates from seed imported from Australia. Isolation of fungi was described in Viljanen-Rollinson et al. (2007). An additional ten Botrytis isolates from Allium plants (Table 3) were obtained from the International Collection of Microorganisms from Plants (ICMP) culture collection http://www.landcareresearch.co.nz/databases/icmp/index.asp. The isolates were maintained on potato-dextrose agar (PDA) slants at 5 °C.
DNA extraction
Fungi were grown on PDA plates for 5–6 days at 21 °C. Mycelium was harvested with a sterilised scalpel and then transferred to a 2-ml screw cap microfuge tube containing 1 g of 2 mm diameter steel beads (Syntech Limited). The mycelium was disrupted for 30 s in buffer with a Mini-Beadbeater-8 (Biospec Product Inc.). Total DNA from fungal isolates was extracted using a cetyl trimethyl ammonium bromide (CTAB) DNA extraction protocol (Russell and Bulman 2005). The DNA precipitation step was modified by the use of 70 % ethanol (Coolong et al. 2008). The final DNA pellet was dissolved in 100 μl of 10 mM Tris–HCl pH 8.5 containing 5 μl of RNase A, and concentration estimated using a NanoDrop™ Spectrophotometer (Thermo Scientific). Final DNA concentrations ranged between 140 ng/μl and 1925 ng/μl.
ITS amplification and sequencing
Details of all primers used in this study are given in Table 1. Locations and relative positions of primers used to amplify and obtain ITS DNA sequence are shown in Fig. 1. Ribosomal ITS PCR amplification was with the ITS5 and ITS26 primer pair as in Bulman and Marshall (1998). Restriction digestion of 5 μl of ITS PCR product was completed in a 10 μl final volume with SphI enzyme (NEB). Digested products were separated in 1 % agarose gels. PCR products were gel-purified and directly sequenced (Macrogen, Korea). Sequences were assembled, and all subsequent bioinformatic analyses were carried out in Geneious (Drummond et al. 2010).

Structure of the predominant ribosomal ITS fragments in the B. aclada and Botrytis isolate I samples. Botrytis isolate I samples have a 1.7 kb insertion in the ribosomal repeat, including the position of a putative group I intron sequence. 18S, 5.8S and 28S are ribosomal genes. The positions of PCR primers used for amplifications are shown above and below sequences with single arrows. SphI = restriction enzyme recognition sites
Specific amplification of part of the large ribosomal ITS insert was performed with the PCR primers ITS5 and BOT_ITSbig_1R. Completion of sequencing of the insert was with the internal primers BOT_ITSbig_1F and BOT_ITSbig_2R (Fig. 1, Table 1). Primers were designed with Primer 3 (Rozen and Skaletsky 2000).
IGS and G3PDH amplification and sequencing
Ribosomal IGS sequences from B. allii, B. byssoidea and B. aclada (Chilvers et al. 2007) were aligned using MUSCLE (Edgar 2004). The forward real-time PCR primer “Chilvers 1F” (Chilvers et al. 2007) was coupled with BOTRY1R to amplify an approximately 900 bp region of the IGS containing variable nucleotides between B. allii and B. aclada. PCR primers for G3PDH amplification and sequencing were from Staats et al. (2005) (Table 1).
PCR reactions were carried out with hot-start Taq polymerases (ABgene or Solis Biodyne) and 1 μl of template DNA (about 50 ng) in each 20 μl reaction. Fragments were amplified using a touchdown PCR protocol (annealing 60 °C × 3 cycles, 58 °C × 3 cycles, 55 °C × 3 cycles and 50 °C × 27 cycles). Extension steps at 72 °C were for 2 min each. PCR products were purified using the QIAquick kit (Qiagen) and directly sequenced (Macrogen, Korea) using the PCR primers. Where necessary, PCR products were cloned in the pCR4-TOPO kit (Invitrogen). Plasmids from insert-containing clones were sequenced with M13F and M13R primers.
DNA sequences generated in this study have been deposited in GenBank under accession numbers JN717253-JN717256 and JX399158-JX399181.
Phylogenies
IGS sequences were aligned with four Botrytis IGS sequences from Chilvers et al. (2007). G3PDH sequences were aligned with a selection of Botrytis sequences, mostly from Staats et al. (2007). Phylogenies were constructed with PHYML using the HKY85 substitution model (Guindon and Gascuel 2003). 1000 bootstraps were completed as a measure of branch support. Phylogenetic analyses were completed in Geneious (Drummond et al. 2010). DNA alignments have been deposited at TreeBASE under accession S13388.
IGS high resolution melt analysis
For high resolution melt analysis (HRMA) identification of IGS sequences, the “Chilvers 1F/1R” real time PCR primers (Chilvers et al. 2007) or a new primer BOTRY2R paired with “Chilvers 1F” (giving an approximately 500 bp product) were used respectively (Table 1). PCR amplifications were of 20 μl volumes containing 10 μl MeltDoctor™ HRMA Master Mix (Applied Biosystems), 0.5 μM each primer, and 10 ng template DNA. PCR reactions were performed using 96-well plates in a PalmCycler (Corbett). Cycling parameters were one cycle of 95 °C for 10 min, 35 cycles of 95 °C for 15 s, 55 °C for 20 s, and 72 °C for 20 s, one cycle of 94 °C for 30 s. The reactions were covered with 20 μl light mineral oil and the plate subject to HRMA analysis in a LightScanner (Idaho Technology Inc.) with a start temperature of 40 °C and an end temperature of 96 °C using auto exposure. Two sample F-tests of variance and t- and z-tests of differences between means were performed on average melting temperature data. A two sample Kolmogorov-Smirnov test was also performed to test the equality of the observed temperature distributions.
The ICMP isolates were used for effectively blind-testing the discriminatory power of the IGS HRMA assay. Isolates identified as Botrytis isolate I (5, 44, 94 and 110) and B. aclada (31, 33 and 41) (Table 2) from our 2005–07 collection were included as standards in the HRMA tests on the ICMP isolates. HRMA tests were duplicated within replicated runs.
Results
ITS sequences
Amplification with the ITS5/26 primer pair yielded a single, approximately 500 bp, DNA product from 75 % of the 48 2005–07 Botrytis DNAs. Sequencing of this ITS product from isolate 15 gave 506 bp of DNA sequence that was identical to GenBank accessions annotated as B. aclada (e.g. Chinese isolate EU093077). The isolate 15 sequence contained two SphI restriction sites with predicted DNA fragments sizes of 137, 200, and 212 bp (including primer sequences) following restriction digestion of the PCR fragments. Surprisingly, eleven of the remaining DNA samples either gave no, or very poor, amplification of the target ITS DNA fragment, despite yielding good amplification of other targets (Fig. 2). Isolate 1 gave weak amplification of the 500 bp ITS fragment, but we nevertheless obtained direct DNA sequence from the sample that was identical to that from isolate 15. Subsequently, we completed SphI digestion for 36 of the New Zealand 2005–07 Botrytis isolates, with all but one (118) giving agarose gel profiles consistent with the presence of two SphI restriction sites (Table 2). Isolate 118 repeatedly gave weak amplification and inconclusive RFLP results.

Agarose gel separation of ribosomal ITS PCR amplifications. The left and centre panels show amplification of ribosomal ITS using the ITS5/26 primer pair from isolates 1, 15, 110, 118, and 137 with 30 s or 3 min extension periods. The longer extension times allowed amplification of > 2 kb ITS fragment in some isolates (arrowed). The right hand panel shows specific amplification of part of the large ribosomal ITS insert from the same isolates using the ITS5/BOT_ITSbig_1R primer pair. Isolate 118 DNA gave consistently poor ITS amplification, but with no evidence of the ribosomal ITS insert. Molecular marker = HyperLadder IV (Bioline)
In PCRs with longer extension times, many of the DNA samples that failed to produce clear amplification of the 506 bp ITS product produced a DNA fragment larger than 2 kb (Fig. 2). These large fragments were directly sequenced from isolates 1 and 26, giving 2252 bp of sequence with no nucleotide differences between the two isolates. Alignment of the 506 bp ITS DNA fragment with the large ITS DNA sequence showed two features (Fig. 1). First, the presence of a 1760 bp insertion was evident within 1–3 bp of the 3′ end of the ITS5 primer position. Second, identical DNA sequence to that of the 506 bp product was present at the 3′ end of the large DNA fragment, but with a variable nucleotide (T vs C) that abolished the first SphI restriction site. BLASTN (Altschul et al. 1990) searches of the 1760 bp inserted sequence showed that approximately 430 bp at the 5′ end had similarity with group I intron sequences from fungi (best hits 2e−146 to Sclerotinia trifoliorum, AY187072). The remainder of the insertion had no similarity to sequences in GenBank based on BLASTN or tBLASTX.
A number of new PCR primers were designed within the 1760 bp insertion (Fig. 1, Table 1). PCR amplification of 187 or 1120 bp DNA fragments confirmed the clear presence of the DNA intron in 12 isolates (Fig. 2, Table 2). One of these isolates (44) was from onion seed originating in Australia.
Later ITS sequencing suggested that, based on BLASTN matches, ICMP 9334 might be B. squamosa, ICMP 14816 Botryotinia porri and ICMP 10226 B. aclada. Among the remaining isolates with B. allii/B. byssoidea-like ITS sequences, four were seen to contain the large intron-like insertion (Table 3). For the remainder of the work, these fungi are referred to as “Botrytis isolate I” to denote the presence of weak ITS amplification and/or ITS intron insertions, their first defining features.
IGS sequences
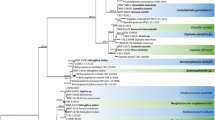
IGS DNA fragments of approximately 950 bp were amplified and sequenced from 13 isolates of the 2005–07 collection (Table 2) and from all 10 ICMP isolates (Table 3). Representative isolates were chosen based on ITS sequencing and ITS-RFLPs. These IGS fragments were directly sequenced with no evidence of double or triple peaks indicative of sequence heterogeneity. DNA alignment and phylogenetics with four GenBank Botrytis sequences from Chilvers et al. (2007) showed that these sequences mostly fell into two groups (Fig. 3). Thirteen sequences clustered with the sequences from B. allii and B. byssoidea while eight identical sequences were the same as that from B. aclada (Chilvers et al. 2007). Within the B. allii/B. byssoidea clade, there were a small number of nucleotide differences that separated groups of New Zealand isolates. For example, ICMP isolates 1682, 1715 and 1716, and isolate 5 from 2005–07 shared a 1 bp deletion and a single nucleotide change. Isolates 1, 2, 94, 110, 129 and ICMP 1719 shared a single nucleotide difference. Perhaps most importantly, ICMP 5601 and ICMP 10259 shared three nucleotide differences with the GenBank B. byssoidea sequence (Fig. 3) (Chilvers et al. 2007). The sequences from ICMP isolates 9334 and 14816 were not closely related to any IGS sequences; ICMP 9334 is on a long branch and is distant sister to the B. allii/B. byssoidea clade (Fig. 3). Similarly, ICMP 14816 is also on a long branch and a distant sister to B. fuckeliana. These two clades form an unresolved tritomy with the B. aclada clade.

PHYML phylogenetic tree constructed from Botrytis ribomomal IGS sequences. New Zealand isolates are shown in bold. Values of greater than 90 % from 1000 bootstraps are shown above nodes. Bar provides a measure of phylogenetic distance
G3PDH sequences
To further confirm the species identifications, we sequenced G3PDH fragments from five 2005–07 isolates (1, 5, 14, 53 and 110) and four of the ICMP isolates. These were a subset of those chosen for IGS sequencing, with emphasis on isolates found to have the most divergent sequences from one another (Fig. 3). The G3PDH PCR fragments from the nine isolates were directly sequenced without evidence of heterogeneity. These sequences were aligned with a broad group of GenBank G3PDH sequences, almost all of which were from Staats et al. (2005). Six identical sequences from New Zealand isolates, two B. aclada and four Botrytis isolate I, were in turn identical to GenBank sequences from B. aclada and (one) B. allii (Fig. 4). The G3PDH sequence from ICMP 10259 was identical to B. byssoidea, that from isolate ICMP 9334 was identical to B. squamosa, while the sequence from ICMP 14816 was most closely related, but not identical, to Botryotinia porri (Fig. 4).

PHYML phylogenetic tree constructed from Botrytis glyceraldehyde-3-phosphate dehydrogenase DNA sequences. New Zealand isolates are shown in bold. Values of greater than 90 % from 1000 bootstraps are shown above nodes. Bar provides a measure of phylogenetic distance
IGS HRMA
HRMA runs carried out on Botrytis isolate I and B. aclada DNAs from the 2005–07 collection, using the Chilvers 1F/1R PCR primers, produced two clear peaks with mean melting temperatures of 74.26 ± 0.07 °C and 75.16 ± 0.07 °C, respectively (Fig. 5). IGS HRMA profiles, produced the same differentiation of isolates as did the ITS restriction patterns and ITS intron presence/absence (Table 2). Two sample F-tests of melting temperature variance showed no significant difference and subsequent t- (T =43.40, P < 0.001) and z-tests (Z = 41.24, P = 0) indicated that the mean melting temperatures for these Botrytis isolate I and B. aclada IGS targets were significantly different from each other. Similarly, a two sample Kolmogorov-Smirnov test (D = 1, P = 0) indicated that the temperature distributions for Botrytis isolate I and B. aclada Chilvers 1F/1R IGS targets were not equivalent. HRMA assays on longer Chilvers 1F/BOTRY2R fragments gave more complicated melting profiles with less clear separation (not shown). HRMA profiles from both primer pairs nevertheless allowed clear separation of isolates with complete agreement between the primer pairs (Table 1).

HRMA detection of IGS variants. a Normalised melt curves obtained during HRMA analysis of Chilvers 1 F/1R IGS fragments for Botrytis isolate I (red) and B. aclada (blue) isolates. b HRMA detection of IGS variants among ICMP isolates. Normalised melt curves obtained during blind test validation of HRMA assay based on Chilvers 1 F/1R primers. Red curve indicates the melt profile for the Botrytis isolate I samples (including reference isolates 5, 44, 94 and 110), the blue curve indicates the melt profile for the B. aclada isolates (including reference isolates 31, 33 and 41), as assigned by the LightScanner software. This software was unable to assign isolates ICMP 9334 and ICMP 14816 to either grouping based on the observed IGS melt temperatures
Results of our assay validation with the Chilvers 1F/1R primer set against the ICMP isolates are shown in Table 3 and Fig. 5. Table 3 shows melting temperature results for the ICMP panel and Fig. 5 shows the melting curves returned for the ICMP DNAs and the reference Botrytis isolate I and B. aclada isolates (Table 2). The average melting temperatures obtained for the reference samples were slightly greater (74.36 ± 0.06 °C for Botrytis isolate I and 75.24 ± 0.07 °C for B. aclada) than the average melting temperatures measured during the runs against the 2005–07 isolates but the differences were of similar magnitude (~0.1 °C) and standard deviations about the means the same. The discriminatory power of the assay was unlikely to be affected. Of the ten ICMP samples, seven returned melt curves similar to the Botrytis isolate I reference samples. These included the two B. byssoidea isolates (5601, 10259), although this was expected because the IGS sequences across the 114 bp IGS fragment were identical between the Botrytis isolate I and B. byssoidea samples. A single isolate, ICMP 10226, had an IGS melting temperature typical of B. aclada. Neither the ICMP 9334 nor 14816 isolates produced IGS melting temperatures that were referable to either Botrytis isolate I or to B. aclada (Table 3, Fig. 5). IGS fragments for HRMA were amplified from several B. squamosa ICMP 9334 template preparations of differing concentration but all returned melting temperatures at least half a degree less than predicted for Botrytis isolate I/B. byssoidea and nearly one-and-a-half degrees less than predicted for B. aclada. By contrast, fragments from B. porri ICMP 14816 melted at temperatures intermediate to those predicted for Botrytis isolate I/byssoidea and B. aclada.
Discussion
Botrytis taxonomy
The between-species variability and multi-copy nature of the ribosomal IGS sequence gene region makes it an attractive target for detecting and quantifying onion-infecting Botrytis (Chilvers et al. 2007). Once our study began, it became apparent that identification of New Zealand Botrytis spp. based on this single gene region would not be possible. Although onion neck rot has been well studied worldwide, the representation of DNA sequences available in GenBank from species causing this disease is not comprehensive; there have previously been few studies where sequences from both ribosomal genes and phylogenetic marker genes (e.g. Staats et al. 2005) were collected from single isolates.
We based our final taxonomic identifications on an iterative series of sequence analyses. Firstly, ribosomal ITS amplification and/or ITS-RFLP, plus some ITS DNA sequencing, was applied to all isolates. Subsequent DNA sequencing of the ribosomal IGS and G3PDH genes from selected isolates was based on the variation seen in the ITS fragment. The results of these sequence analyses as well as IGS HRMA assays (on all isolates) were consistent with one another and indicated that we had identified the full range of isolates and species within the available Botrytis collections. From this combined data, the majority of the recent 2005–07 Botrytis isolates were determined to be B. aclada. This stands in contrast to much researcher (e.g. Viljanen-Rollinson et al. 2007) and industry assumption that onion neck rot in New Zealand is associated with B. allii.
Among isolates stored in the ICMP, B. byssoidea, B. squamosa and Botryotinia porri were identified. In all cases, these Botrytis spp. have been reported to be in New Zealand, but these are the first molecular confirmations of their presence. It is also notable that the original (although often tentative) species designations presented in the ICMP database bore a poor resemblance to the species identities as revealed here. In the absence of easily available taxonomic expertise based on fungal morphology, DNA based identifications of Botrytis isolates will enhance our ability to study the ecology of these fungi in New Zealand.
Identification to species level for the remaining 12 2005–07 isolates and five of the culture collection isolates was more ambiguous. Although these isolates had ITS sequences matching that in B. allii/B. byssoidea, all but one contained a distinctive intron insertion at the 5′ end of this ITS fragment. Several were also found to contain low amounts of a second ITS fragment characteristic of B. aclada. The detection of two ribosomal ITS sequences in these isolates could be consistent with the predicted hybrid nature of the B. allii genome (Nielsen and Yohalem 2001; Yohalem et al. 2003) although incomplete homogenisation of the ribosomal repeats has not been detected in this species (Nielsen and Yohalem 2001). Low levels of one type of ITS sequence might not have been apparent via direct sequencing or PCR-RFLP approaches in earlier studies, and might only be obvious here due to the large size of the predominant ITS fragment.
Group I introns are a common feature in fungal ribosomal genes (Holst-Jensen et al. 1999), including at sites bordering the ribosomal ITS (e.g. Johnston and Park 2007). These introns are characterised by insertion at conserved sites in the ribosomal genes and evidence of possible horizontal transfer between members of the Sclerotiniaceae has been presented (Holst-Jensen et al. 1999). Given the similarity of the Botrytis isolate I insertions to those from Sclerotinia trifoliorum, it is possible to speculate that this represents a horizontal transfer event between members of the Sclerotiniaceae, especially given that Botrytis spp. and Sclerotinia cepivorum (that causes onion white rot disease) are often found in close proximity on onion bulbs. Group I introns are also increasingly used as diagnostic markers for fungal lineages (e.g. Garrido-Jurado et al. 2011). Njambere et al. (2010) have recently utilised a PCR assay to detect an intron adjacent to the ribosomal ITS1 spacer in Sclerotinia trifoliorum but not in S. sclerotiorum. This has many similarities to our intron-detection assays presented here. Wider and more in depth sampling of Botrytis samples will be needed to determine the origin and prevalence of this intron and of Botrytis isolate I. For example, how geographically widespread are the Botrytis isolate I fungi, given that one of our isolates was obtained from onion seed imported from Australia?
Do these molecular aspects of the Botrytis isolate I fall within global diversity of B. allii? Ribosomal IGS sequences placed these isolates close to B. allii and B. byssoidea, whereas their G3PDH sequences were identical to B. aclada (Staats et al. 2005). A lack of heterogeneity in the direct sequences from the G3PDH gene was inconsistent with the allodiploid B. allii (Staats et al. 2005). Thus the Botrytis isolate I sequences did not conform to the expected pattern for B. aclada, B. byssoidea or B. allii. Moreover, to the best of our knowledge, the presence of intron-type insertions in ribosomal DNA fragments has not been reported in B. allii or any other onion neck rot-causing Botrytis spp. (Nielsen et al. 1999; Nielsen et al. 2001). Taxonomic resolution of Botrytis isolate I awaits further work, perhaps using NEP1 and NEP2 gene sequences which are quite distinctive between Botrytis spp. (Staats et al. 2007). In addition to Botrytis isolate I and the recently described Botrytis sinoallii from onion crops in China (Zhang et al. 2010), a North American B. aclada isolate MUCL3106 did not display exactly the same pattern of DNA sequences found among European B. aclada isolates (Staats et al. 2005). Given these findings, we contend that genome evolution among the wider group of onion-infecting Botrytis spp. warrants further study, and certainly collection of multiple gene sequences from isolates sourced over a wider geographical selection of environments. Lastly, based on these studies, it remains an open question whether the allodiploid B. allii has ever been present in New Zealand.
The results presented raise questions about the influence of management practices upon the species composition of onion-infecting Botrytis in New Zealand. Our identification of predominantly B. aclada in 2005–07 can be compared with two Northern Hemisphere studies of 29 (Nielsen et al. 2001) and 469 (Chilvers and du Toit 2006) isolates respectively, which showed somewhat more equal numbers of B. allii and B. aclada involved in onion neck rot disease. In contrast, a study of 23 Australian isolates found that all were B. allii with the exception of a single B. aclada isolate (Chilvers et al. 2004). Our data suggest that a shift in the distribution of Botrytis spp. on onions in New Zealand has occurred since 2002. While our pre-2003 sample set was small, most isolates collected from Allium plants were the Botrytis isolate I or B. byssoidea, with only a single B. aclada. Between 2005 and 2007, Botrytis isolate I was infrequently collected relative to B. aclada, and B. byssoidea was not found at all.
Species-specific diagnostic assays
To more rapidly identify Botrytis spp., we adopted the technique of HRMA on IGS sequences. HRMA is based upon the slow melting of double stranded DNA and measurement of fluorescence changes when a DNA bound dye is released. The characteristic melting peak (when the change in fluorescence is maximal) of each DNA sequence is dependent on the fragment length and GC content (Mackay and Wittwer 2011). By applying HRMA after amplification of the 114 bp Chilvers IGS fragment, we developed a new assay that rapidly separated B. aclada from Botrytis isolate I and B. byssoidea. This application of IGS HRMA gave a congruent series of results to those produced from ITS-RFLP, specific PCR for the ribosomal intron insert, and DNA sequencing from selected isolates. Such HRMA techniques offer advantages over available PCR-RFLP because they are closed tube assays requiring only brief post-amplification processing, are cheap, rapid and simple to use (Vossen et al. 2009). HRMA allows differentiation of multiple alleles from one PCR reaction and can also be used to confirm species in a post-amplification melt following SYBR green quantitative PCR.
Distinguishing distantly related sequences rather than single nucleotide polymorphisms is still a relatively uncommon application of HRMA, but is an area of current development (Mader et al. 2008; Talmi-Frank et al. 2010; Winder et al. 2011). In this case, the 114 bp fragment that contained nine nucleotide differences between B. aclada and the Botrytis isolate I/B. byssoidea gave a single peak melting profile that was readily interpreted, whereas a longer, overlapping 506 bp fragment of the IGS gave a more complicated melting profile. This longer fragment presumably encompassed multiple melting domains and the advantages of shorter amplicons for HRMA have been previously described (Vossen et al. 2009).
The HRMA assay did not distinguish between Botrytis isolate I and B. byssoidea because these have identical sequences across this 114 bp IGS fragment. However, our 2005–07 collection of onion-infecting Botrytis suggested that B. aclada was the predominant species in New Zealand with no B. byssoidea detected. Botrytis byssoidea has also infrequently been reported to cause onion neck rot internationally (Chilvers and du Toit 2006). Other regions of the ribosomal IGS could be targeted to differentiate B. byssoidea from other Botrytis spp., should this be desired. DNA samples from overseas isolates of onion-associated Botrytis, including verified B. allii, are being sought in order to test the limits of this assay.
Whether the HRMA assay will be applicable in field samples will require further research. HRMA does seem to be sensitive to variable DNA concentration and quality, and some studies have suggested it is less robust than other molecular diagnostic techniques (Bass et al. 2007; Mackay and Wittwer 2011). In addition to the HRMA assays, the PCR primer pairs targeting the Botrytis isolate I intron sequence also represent a new assay for the specific detection of these isolates.
The tools developed in this study have provided substantial new insights into the ecology of onion-infecting Botrytis spp. in New Zealand. Further collection of Botrytis is underway and we are developing complementary tools that will address emerging questions about fungicide resistance in these species.
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., & Lipman, D. J. (1990). Basic local alignment search tool. Journal of Molecular Biology, 215, 403–410.
Bass, C., Nikou, D., Donnelly, M. J., Williamson, M. S., Ranson, H., Ball, A., et al. (2007). Detection of knockdown resistance (kdr) mutations in Anopheles gambiae: a comparison of two new high-throughput assays with existing methods. Malaria Journal, 6. doi:10.1186/1475-2875-6-111.
Bulman, S. R., & Marshall, J. W. (1998). Detection of Spongospora subterranea in potato tuber lesions using the polymerase chain reaction (PCR). Plant Pathology, 47, 759–766.
Chilvers M. I. & du Toit L. J. (2006). Detection and identification of Botrytis species associated with neck rot, scape blight, and umbel blight of onion. Plant Health Progress, 1–13.
Chilvers, M., Pethybridge, S. J., Hay, F. S., & Wilson, C. R. (2004). Characterisation of Botrytis species associated with neck rot of onion in Australia. Australasian Plant Pathology, 33, 29–32. doi:10.1071/ap03077.
Chilvers, M. I., du Toit, L. J., Akamatsu, H., & Peever, T. L. (2007). A real-time, quantitative PCR seed assay for Botrytis spp. that cause neck rot of onion. Plant Disease, 91, 599–608. doi:10.1094/pdis-91-5-0599.
Coolong, T. W., Walcott, R. R., & Randle, W. M. (2008). A quantitative real-time polymerase chain reaction assay for Botrytis aclada in onion bulb tissue. Hortscience, 43, 408–413.
Drummond A. J., Ashton B., Cheung M., Heled J., Kearse M., Moir R., et al. (2010). Geneious v5.0. Available from http://wwwgeneiouscom,
du Toit, L. J., Derie, M. L., & Pelter, G. Q. (2004). Prevalence of Botrytis spp. in onion seed crops in the Columbia basin of Washington. Plant Disease, 88, 1061–1068.
Edgar, R. C. (2004). MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics, 5, 1–19. doi:10.1186/1471-2105-5-113.
Garrido-Jurado, I., Marquez, M., Ortiz-Urquiza, A., Santiago-Alvarez, C., Iturriaga, E. A., Quesada-Moraga, E., et al. (2011). Genetic analyses place most Spanish isolates of Beauveria bassiana in a molecular group with word-wide distribution. BMC Microbiology, 11. doi:10.1186/1471-2180-11-84.
Guindon, S., & Gascuel, O. (2003). A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Systematic Biology, 52, 696–704. doi:10.1080/10635150390235520.
Holst-Jensen, A., Vaage, M., Schumacher, T., & Johansen, S. (1999). Structural characteristics and possible horizontal transfer of group I introns between closely related plant pathogenic fungi. Molecular Biology and Evolution, 16, 114–126.
Johnston, P. R., & Park, D. (2007). Revision of the species of Rhytismataceae reported by Spegazzini from South America. Boletin de la Sociedad Argentina de Botanica, 42, 87–105.
Lorbeer, J. W., Seyb, A. M., de Boer, M., & van den Ende, J. E. (2004). Botrytis species on bulb crops. Springer: Dordrecht.
Mackay, J. F., & Wittwer, C. T. (2011). High-resolution melting analysis. In N. Oswald & S. Kennedy (Eds.), PCR troubleshooting and optimization: The essential guide (pp. 201–206). Wymondham: Caister Academic.
Mader, E., Lukas, B., & Novak, J. (2008). A strategy to setup codominant microsatellite analysis for high-resolution-melting-curve-analysis (HRM). BMC genetics, 9, 69.
Maude, R. B., & Presly, A. H. (1977). Neck rot (Botrytis allii) of bulb onions I. Seed-borne infection and its relationship to the disease in the onion crop. Annals of Applied Biology, 86, 163–180.
Nielsen, K., & Yohalem, D. S. (2001). Origin of a polyploid Botrytis pathogen through interspecific hybridization between Botrytis aclada and B.byssoidea. Mycologia, 93, 1064–1071.
Nielsen K., Justesen A. F. & Yohalem D. S. (1999) PCR based detection of latent infections of Botrytis aclada Fres. in onion bulbs. In: Proceedings of the meeting, Mass scale diagnosis of plant pathogens by nucleic-acid amplification methodologies, held 9–10 July 1998 in Faro, Portugal.
Nielsen, K., Justesen, A. F., Jensen, D. F., & Yohalem, D. S. (2001). Universally primed polymerase chain reaction alleles and internal transcribed spacer restriction fragment length polymorphisms distinguish two subgroups in Botrytis aclada distinct from B. byssoidea. Phytopathology, 91, 527–533.
Njambere E. N., Attanayake R. N. & Chen W. (2010). Applications of molecular markers and DNA sequences in identifying fungal pathogens of cool season grain legumes. Molecular identification of fungi / edited by Youssuf Gherbawy, Kerstin Voigt (pp. 79–91). Springer
Rozen, S., & Skaletsky, H. (2000). Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol, 132, 365–386.
Russell, J., & Bulman, S. (2005). The liverwort Marchantia foliacea forms a specialized symbiosis with arbuscular mycorrhizal fungi in the genus Glomus. New Phytologist, 165, 567–579. doi:10.1111/j.1469-8137.2004.01251.x.
Shirane, N., Masuko, M., & Hayashi, Y. (1989). Light microscopic observation of nucleic and mitotic chromosomes of Botrytis species. Phytopathology, 79, 728–730.
Staats, M., van Baarlen, P., & van Kan, J. A. L. (2005). Molecular phylogeny of the plant pathogenic genus Botrytis and the evolution of host specificity. Molecular Biology and Evolution, 22, 333–346. doi:10.1093/molbev/msi020.
Staats, M., van Baarlen, P., Schouten, A., & van Kan, J. A. L. (2007). Functional analysis of NLP genes from Botrytis elliptica. Molecular Plant Pathology, 8, 209–214. doi:10.1111/j.1364-3703.2007.00382.x.
Stewart, A., & Franicevic, S. C. (1994). Infected seed as a source of inoculum for Botrytis infection of onion bulbs in store. Australasian Plant Pathology, 23, 36–40.
Talmi-Frank, D., Nasereddin, A., Schnur, L. F., Schonian, G., Toz, S. O., Jaffe, C. L., et al. (2010). Detection and identification of old world Leishmania by high resolution melt analysis. PLoS Neglect Trop Dis, 4. doi:10.1371/journal.pntd.0000581.
Viljanen-Rollinson, S., Marroni, M., & Butler, R. (2007). Reduced sensitivity to carbendazim in isolates of Botrytis allii. New Zealand Plant Protection, 60, 108–113.
Vossen, R., Aten, E., Roos, A., & den Dunnen, J. T. (2009). High-resolution melting analysis (HRMA)-more than just sequence variant screening. Human Mutation, 30, 860–866. doi:10.1002/humu.21019.
Winder, L., Phillips, C., Richards, N., Ochoa-Corona, F., Hardwick, S., Vink, C. J., et al. (2011). Evaluation of DNA melting analysis as a tool for species identification. Methods in Ecology and Evolution, 2, 312–320. doi:10.1111/j.2041-210X.2010.00079.x.
Yohalem, D. S., Nielsen, K., & Nicolaisen, M. (2003). Taxonomic and nomenclatural clarification of the onion neck rotting Botrytis species. Mycotaxon, 85, 175–182.
Zhang, J., Zhang, L., Li, G.-Q., Yang, L., Jiang, D.-H., Zhuang, W.-Y., et al. (2010). Botrytis sinoallii: a new species of the grey mould pathogen on Allium crops in China. Mycoscience, 51, 421–431. doi:10.1007/s10267-010-0057-4.
Acknowledgment
Research was supported by the Ministry of Science and Innovation Future Vegetables (C02X0701) programme. We thank Maureen Fletcher for generously supplying ICMP isolates. Jan van Kan, Peter Johnston and Martin Chilvers provided generous advice regarding fungal diagnostics. An anonymous reviewer greatly helped construction of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Khan, M.I., Marroni, V., Keenan, S. et al. Enhanced molecular identification of Botrytis spp. from New Zealand onions. Eur J Plant Pathol 136, 495–507 (2013). https://doi.org/10.1007/s10658-013-0182-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10658-013-0182-y