
Abstract
Sugarcane yellow leaf virus (SCYLV) is the causal agent of the sugarcane disease Yellow leaf (YL), which was first reported in Hawaii. The presence of SCYLV was detected by tissue blot immunoassay and the Hawaiian sugarcane cultivars fell into susceptible cultivars (with SCYLV) and resistant cultivars (without SCYLV). RT-PCR showed recently that the resistant cultivars also contain the virus, however with a 100-fold lower virus titre than in the susceptible cultivars. SCYLV is present as whole genome (6 kb) or as two subgenomic sequences of 2.4 and 1.0 kb. Virus preparations from three Hawaiian cultivars (one resistant and two susceptible) were fully sequenced and the sequences were aligned to published, full and partial sequences. The phylograms corroborate previous findings that the so-called YLS-segment, which codes for the coat protein, shows the least genetic diversity, whereas the other sequence fragments A–D, representing the ORFs 0–5, expressed a two-fold higher diversity. The Hawaiian SCYLV-strains clustered together next to the Peru strain, apart from the BRA-strains and well apart from the REU-strains. We propose that the Hawaiian SCYLV should be considered as an independent group together with the Peru strain and known as HAW-PER. The sequences from the two susceptible cultivars had a deletion of 48 to 54 nt in ORF1, which codes for the gene silencing suppressor and a RNA-dependent RNA-polymerase. It is speculated that this deletion is important for the high proliferation rate of the virus in the susceptible plants.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The sugarcane disease Yellow leaf (YL) was first reported from plantations on two Hawaiian islands (Schenck 1990) and described as Yellow leaf syndrome. Few years later similar symptoms were observed in mainland US (Comstock et al. 1994) and Brazil together with dramatic yield losses (Vega et al. 1997). The symptoms are characterized by the yellowing first of leaf midribs followed by that of the entire leaf blade and internode shortening, which leads to a bushy appearance of the green leaf top. Borth et al. (1994) reported a dsRNA-virus in diseased plants. Later, a luteovirus (ss+ RNA) could be unequivocally identified as the causal agent of Yellow leaf (Vega et al. 1997) and it was named Sugarcane yellow leaf virus (SCYLV). The Hawaiian cultivars were classified according to the presence of SCYLV into susceptible and resistant cultivars (Schenck and Lehrer 2000), based on the observation that all plants of susceptible cultivars contained SCYLV when tested by tissue blot immunoassay (TBIA), whereas plants from resistant cultivars appeared virus-free. The strength of YL-symptom expression was correlated (though not strictly) to the presence of SCYLV (Lehrer and Komor 2008). Recent data obtained by PCR indicated that the resistant Hawaiian cultivars also contained SCYLV although at much lower titre (Zhu et al. 2010).
Sequence analyses revealed that some regions of SCYLV genome are closely related to Barley yellow dwarf virus and others similar to the Potato leaf roll virus, which suggested that SCYLV may be a recombination product of a Polerovirus and an Enamovirus (Moonan et al. 2000; Smith et al. 2000). SCYLV-strains from different American countries were characterized by fingerprinting and partial sequence analysis and a Colombian strain was postulated as a founder strain of SCYLV (Moonan and Mirkov 2002). Later Abu Ahmad et al. (2006, 2007) compared 60 SCYLV-preparations from almost all sugarcane-growing countries (including Colombia) by diagnostic PCR-reactions or by partial sequencing. They grouped the virus into 2 major strains, one called REU which is found mainly in Reunion Island, the other BRA-PER, which is found in North and South America. SCYLV from Hawaiian cultivars were not among that study, although the disease and SCYLV were first detected in Hawaii. Yet some SCYLV-preparations had a relationship to Hawaii, because a SCYLV preparation from cultivar R570 which was grown in the collection of the Hawaiian sugarcane breeding station, contained the BRA-strain and not the REU-strain which exists in R570 which is grown in Réunion. The Hawaiian cultivars H32-8560 and H50-7209, which were exported to Peru in 1981, were found to be infected with the PER strain, which is closely related to but not identical with the BRA-strain (Abu Ahmad et al. 2006). Therefore, it was reasonable to assume that the sugarcane plantations of the Hawaiian Islands are infected by BRA and/or PER strains of SCYLV, however, direct evidence for this assumption is lacking.
The objective of this study was to sequence SCYLV from susceptible and resistant Hawaiian cultivars and to determine their phylogenetic relationships to already reported SCYLV clusters. In addition, we wanted to assure that the so-called YLS-segment of SCYLV, which is considered as a diagnostic sequence for all SCYLV-strains (Comstock et al. 1998; Abu Ahmad et al. 2006, 2007), is a reliable sequence part for quantification of SCYLV in susceptible and resistant cultivars by real-time PCR (Zhu et al. 2010).
Material and methods
Plant material and aphids
Cultivars H73-6110, H87-4319, H78-4153, H65-7052, H78-7750, and H87-4094 were obtained from the Hawaii Agriculture Research Center, Aiea, Hawaii, USA. A virus-free line of the cultivar H87-4094 was produced by meristem tip tissue culture and was provided by Dr. A. Lehrer, Honolulu. In addition, cultivars C1051-73, JA-605 and CP52-43, were obtained from Cuba through Isabel Medina Borges, Habana. The plants were grown in the greenhouse at Bayreuth University at 24°C with a 12-h photoperiod and propagated 1-2 times per year from cuttings.
Aphids Melanaphis sacchari were collected from sugarcane at Hawaiian Agriculture Research Center, Kunia, Hawaii, USA
Isolation of RNA, RT-PCR and northern blot for detection of SCYLV
RNA was extracted and purified from the top visible dewlap leaf as previously described (Comstock et al. 1998; Sambrook and Russell 2001; Komor et al. 2010).
RNA was extracted from aphids by the same protocol as described above with RNA extraction buffer (4 M guanidine thiocyanate (Sigma-Aldrich, Chemie GmbH, Munich, Germany), 25 mM sodium citrate, pH 7.5% Sarkosyl (Sigma-Aldrich) and 2 M sodium acetate pH 4.0).
RT-PCR was used to test the presence of SCYLV in the leaf samples of 9 Sugarcane cultivars and one aphid sample using diagnostic primers YLS111 and YLS462 (Comstock et al. 1998). The RNA was reverse transcribed using RevertAid H Minus First Strand cDNA Synthesis Kit (Fermentas GmbH, Leon-Rot Germany), primed with 50 pmol of YLS462 by following the manufacturer’s protocol in a PCR machine (PTC 100 Peltier Thermal Cycler, MJ Research, Global Medical Instrumentation, Inc, Ramsey, Minnesota, USA.). The RT-PCR reaction was performed in 25 μl containing 1 μl cDNA, 2.5 μl of 10× PCR buffer containing 15 mM MgCl2, 0.5 μl of 10 mM dNTP mix, 10 pmol each of forward and reverse primers (YLS111 and YLS462), 1 unit of a polymerase with proofreading activity (Pfu ): Taq polymerase (5:1) (Stratagene, Waldbronn, Germany), and sterile milliQ water added up to the final volume of 25 μl. This PCR programme was performed with initial denaturation at 94°C for 4 min, 10 cycles of 94°C for 30 s, 62°C for 2 min, 72°C for 1.5 min, and 30 cycles of 94°C for 30 s, 62°C for 30 s, 72°C for 1.5 min with a final 72°C extension for 7 min. The primer pairs located in the different ORFs of SCYLV genome are listed in Table 1 and Fig. 1.

RT-PCR for SCYLV in 10 Hawaiian cultivars, 3 Cuban cultivars and in viruliferous Melanaphis sacchari. RNA from sugarcane leaves and from aphids was extracted, transcribed to cDNA and amplified with diagnostic primers (YLS111 and YLS462) by RT-PCR. H73-6110 and H87-4094 are susceptible, H65-7052 intermediately susceptible and H78-4153, H87-4319 and H78-7750 resistant cultivars. The virus-free clone of H87-4094 is used as a negative control, in addition 3 cultivars obtained from Cuba (C1051-73, JA-605 and CP52-43) were tested. The PCR products were electrophoresed on 1% agarose gel and stained with ethidium bromide. M: DNA molecular size marker, loading control 25S rRNA, 108 bp
Northern blots were prepared according to Sambrook and Russell (2001) with 10 μg of intact RNA isolated from sugarcane leaves.
Genome fragment amplification
Genome fragments A–D, YL0, YL1, YL5 and YLS were amplified from reverse-transcribed RNA preparations as described above. The partial ORFs 0 and 1 (fragment A) was amplified from three cultivars (H73-6110, H87-4319 and H87-4094). The PCR program for the amplification of partial ORFs 0 and 1 with the primers ORF1START and 160R.640R was 94°C for 5 min, 10 cycles of 94°C for 30 s, 62°C for 2 min, 72°C for 4 min, and 30 cycles of 94°C for 30 s, 62°C for 30 s, 72°C for 4 min with a final 72°C extension for 15 min. PCR program performed with the primers oFM323 and oFM359 was 94°C for 5 min, 10 cycles of 94°C for 30 s, 58°C for 2 min, 72°C for 4 min, and 30 cycles of 94°C for 30 s, 58°C for 30 s, 72°C for 4 min with a final 72°C extension for 15 min. Partial sequence of ORF2, ORF5 and complete sequence of ORF3 and ORF4 (Fragment C) were amplified with three cultivars (H73-6110, H87-4319 and H87-4094). The RT-PCR program performed with primers B FOR and B REV was 94°C for 5 min, 10 cycles of 94°C for 30 s, 62°C for 2 min, 72°C for 4 min, and 30 cycles of 94°C for 30 s, 62°C for 30 s, 72°C for 4 min with a final 72°C extension for 15 min.
The partial sequence of ORF5 (fragment D) was amplified from RNA isolated from the cultivars H73-6110, H87-4319 and H87-4094 with 104R.623R and 3′PRIME2 primer pair. The RT-PCR program was 94°C for 5 min, 10 cycles of 94°C for 30 s, 62°C for 2 min, 72°C for 4 min, and 30 cycles of 94°C for 30 s, 62°C for 30 s, 72°C for 4 min with a final 72°C extension for 15 min.
PCR reaction for amplification of the gap in ORF1 in SCYLV genome (fragment YL1) was performed with the YL1FOR and YL1REV primer pair to cover the non-sequenced region. The PCR programme was the same as for primers for YLS. The amplification of the region in ORF0 in SCYLV genome (fragment YL0) was performed with primer ORF0 FOR and ORF0 REV and the PCR programme was the same as mentioned above with primer YLS. Additionally, the fragment YL5 was amplified with primer ORF5 FOR and ORF5 REV and the PCR programme was the same as mentioned above with primer YLS.
The RT-PCR was performed with internal control, 25S rRNA, as a reference gene to normalize gene expression level and to evaluate the integrity of cDNA. Furthermore, the primer sets were optimized using semi qPCR with different numbers of PCR-cycles.
A 15 μl aliquot of each amplified product was analysed by electrophoresis on 1% agarose gels stained with ethidium bromide.
Cloning and sequencing of RT-PCR products
Twenty-five amplicons of all viral fragments, derived from independent RT-PCR reactions were cloned in the pGEM®-T Easy Vector System (Promega, Mannheim, Germany) and were transformed into the competent E. coli DH5α strain. The recombinant DNA clones containing the inserts were purified using the Pure yield™. Plasmid Miniprep System (Promega, Mannheim, Germany). The selected clones were sequenced by primer walking using M13 sequencing primers and internal primers specific for each of the fragments A, B, C, and D in the DNA Analytics Core Facility at the University of Bayreuth. One clone per amplicon was sequenced and used for alignment and phylogenetic analyses.
Alignment of sequences and construction of phylogenetic trees
Multiple sequence alignments of nucleotide or deduced amino acid sequences were aligned using CLUSTAL W applying the Dayhoff PAM 250 matrix (Thompson et al. 1994) and were optimized manually. Phylogenetic reconstructions were performed using Geneious program, version 4.8.5 (www.geneious.com). Trees of subgenomic fragments were constructed by Genious’ treebuilder algorithm using the UPGMA method. Data sets were bootstrapped (1,000 replicates) to assess the confidence values of the phylogenetic trees, and bootstrap values <50% were omitted. A phylogenetic tree using the entire genomic sequences of published viruses and of the almost complete genomic sequences of the three virus isolates of this study was reconstructed using Geneious’ PhyML plugin (Guindon and Gascuel 2003) (GTR substitution model; bootstrap: 1,000 replicates). The GenBank accession numbers of the sequences determined here and those used for phylogenetic analysis are listed in Table 2.
Results
Detection of SCYLV in Hawaiian cultivars using RT-PCR and northern blot analysis
RNA was isolated from ten cultivars (seven from Hawaii and three from Cuba) and used for cDNA synthesis and SCYLV detection. The diagnostic primers YLS111 and YLS462 were used in the PCR reactions (Abu Ahmad et al. 2006). A virus-free line of cultivar H87-4094 was generated by meristem tip culture and used as negative control. As expected, two susceptible Hawaiian cultivars (H73-6110 and H87-4094) showed strong amplification products of SCYLV of the expected size, while the virus-free line H87-4094Vf showed no PCR product (Fig. 1). The three resistant cultivars (H87-4319, H78-4153 and H78-7750) showed a quite different amplification patterns. While H78-4153 and H78-7750 generated a weak band corresponding to the correct size, H87-4319 expressed a relatively strong amplification product. The three Cuban cultivars (C1051-73, JA-605 and CP52-43) showed weak bands of SCYLV, similarly aphids feeding on cv. H87-4094. Using different numbers of RT-PCR cycles, the differences in virus titre between susceptible and resistant cultivars could be estimated to be at least 100-fold (data not shown).
Northern blot analysis revealed an accumulation of SCYLV at high level in the lines H73-6110, H87-4094 and H87-4319 (Fig. 2). The cultivars H78-4153, H65-7052 and H78-7750 showed no signal indicating no virus or a low titre below the detection threshold. The genome of SCYLV apparently contained genomic RNA (gRNA) and two subgenomic RNAs (sgRNAs). The estimated molecular size of the gRNA was 6.0 kb, the estimated molecular sizes of the sgRNAs were 1.0 and 2.4 kb.

Northern blot of SCYLV from Hawaiian sugarcane. The RNA gel blot was probed with DIG-labeled SCYLV-probe covering the YLS-sequence and detected with anti-digoxigenin-AP and CDP-Star ready-to-use and visualized with a chemilux CCD camera (Intas, Göttingen, Germany). The apparent size of the hybridization signals was deduced from RNA molecular size markers (not shown), loading control 25S rRNA visualized by probe hybridization
Fragment amplification of SCYLV isolates and phylogenetic relationship to published SCYLV-isolates
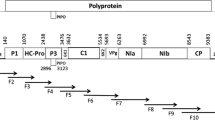
SCYLV is a ss+-RNA virus with 5895-5899 nucleotides organized in six open reading frames (ORFs 0–5) (Fig. 3). Eight primer pairs for amplification of fragments YLS and A-D were designed (Table 1) for the six open reading frames. Complete sequences of SCYLV from 3 Hawaiian cultivars and several partial sequences (accession numbers: GU570004, GU570005, GU570006, GU570007, GU570008, GU570009, GU570010) were obtained.

SCYLV-genome organization, functional open reading frames and positions of amplified SCYLV fragments. Aphid transmission f. = putative aphid transmission factor, CP = capsid protein, MP = putative movement protein, ORF: open reading frame, RdRp = RNA-dependent RNA-polymerase, UTR: untranslated region, F.U.: function unknown
The Hawaiian amplicons were used for phylogenetic analysis together with sequences from the GenBank data base (http://www.ncbi.nlm.nih.gov) (Table 2). Since the SCYLV-genome is a recombination product of two Luteoviridae viruses, the phylogenetic relationship of fragments A–D and YLS were separately constructed to visualize possible sequence segments, where the Hawaiian strains may have diverged from other strains.
The nucleotide sequences of the YLS-region were obtained from 44 SCYLV isolates. The Hawaiian sequences were assembled in cluster YLS1 (Fig. 4a). The resistant cultivar (Haw87-4319) showed 100% sequence identity to YLS from Brazil, Florida, China and Réunion. the YLS-sequence from the two susceptible Hawaiian cultivars (Haw73-6110 and Haw87-4094) exhibited close sequence similarity to those from Australian and Peru. Only SCYLV from Indian cultivars (and one Colombian) were clustered separately from all other YLS-sequences (cluster YLS2 in Fig. 4a). The deduced amino acid sequences of the capsid protein (CP) obtained from all the isolates expressed almost identical amino acid sequences (97–100%, data not shown).

Phylogenetic trees based on nucleotide sequence alignments of SCYLV-isolates including the Hawaiian isolates (arrows). The trees were constructed with Geneious’ tree builder algorithm and UPGMA method (a–e) or with the PhyML algorithm (e); Scale bars: substitutions per site. a Phylogram of fragment YLS (351 nt), b Phylogram of fragment A (1620 nt), c Phylogram of fragment B (1211 nt), d Phylogram of fragment C (1879 nt), e Phylogram of fragment D (1299 nt), f Phylogram of published complete genomes
Forty-three virus isolates were used in phylogenetic analysis of fragment A (comprising partial ORFs 0 and 1). The sequences were distributed into three major groups (Fig. 4b). Cluster A1 contained 19 SCYLV sequence isolates from many origins (USA, Brazil, China, Australia etc.), the Hawaiian isolates were together in cluster A2 and the Peru isolates (A3) appeared similarly close to A1 (BRA) and Hawaiian isolates (A2). The sequence identity among Hawaiian isolates ranged between 91.2% and 98.9%. The majority of Réunion virus strains were clustered into group A3. (MUS1 seemed to be unique not fitting in any of the above groups.)
Thirty-seven sequences of fragment B (partial ORF2) were classified into three clusters (Fig. 4c), the Hawaiian virus isolates (Haw73-6110, Haw87-4319 and Haw87-4094) clustered in B1 as unique subgroup with a sequence identity between 89.5% and 90.3%. The Réunion isolates were grouped together in cluster B2, cluster B3 contained two Cuban virus isolates (CUB-YL1 and CP52-43) with 98.7% identity.
The sequences of 21 isolates of fragment C, which covers ORFs 3 and 4 and parts of ORFs 2 and 5, were assembled into 5 clusters (Fig. 4d). Nine SCYLV-isolates from different geographical origins (Brazil, USA etc.) were grouped in cluster C1 next to the Hawaiian isolates in cluster C2, C3 contained the PER-isolates, C4 the REU-isolates and cluster C5 included only Colombia isolates.
Thirty-eight SCYLV isolates of fragment D which is related to the putative aphid transmission factor, were grouped into three clusters (Fig. 4e). Cluster D1 contained subgroups from various origins of SCYLV isolates (e.g. Florida, Brazil, India in one subgroup, isolates REU40 and REU42 in another subgroup). Hawaiian isolates were assembled in cluster D2 as unique subgroup, the majority of Réunion virus isolates was clustered in D3 together with two isolates from Mauritius.
The phylogenetic analysis of the SCYLV partial sequences constantly yielded 3 clusters: (1) a group comprising strains from Brazil, USA, China, India and several other countries (tentatively group BRA according Abu Ahmad et al., 2006), (2) a group exclusively for most strains from Réunion (group REU) and (3) a group with the Hawaiian and the Peru strains. For this group we propose the name HAW-PER. In fragment C a Colombian cluster, in fragment B a Cuban cluster showed up, separated from the other strains. Phylogenetic analysis of 14 complete sequences of SCYLV genome also exhibited three groups: Group HAW-PER included two subgroups with the Hawaiian and Peru isolates and a strain from Florida with bootstrap value 89%, group 2 (BRA) formed by isolates from various origins (Brazil, China, India and USA) and group 3 with the REU strains. Thus the whole genome confirms the results already obtained from the alignment of partial sequences which are available in much larger numbers.
The genome sequences were analysed by Recco Nucleotide alignment Geneious fasta to test whether the Hawaiian strains may have evolved through recombination of so-far sequenced SCYLV-strains. Only one possible recombination event was noticed, namely at nucleotide position 4375-4389 between H87-4094 and H73-6110 to yield H87-4319. The recombination would have “saved” 7 mutational events (data not shown).
Deletion/insertion in ORF1
The nucleotide sequences from susceptible cultivars H78-6110 and H87-4094 and the resistant cultivar H87-4319 showed a lack of 48 to 54 nucleotides in the susceptible cultivars (Fig. 5a). A 51 nt deletion was detected in fragments A and B of cultivar H87-4094 corresponding to nucleotides 1686 to 1736 of SCYLV (NCBI accession NC_000874, Moonan et al. 2000). In contrast, a 48 nt deletion was detected in two independent A fragments obtained from cultivar H73-6110 corresponding to nucleotides 1686 to 1733 of SCYLV and a 54 nt deletion corresponding to nucleotides 1681 to 1734 in fragment B. Since these deletions were detected in independent amplification products of these two sugarcane cultivars, they did most likely not result from amplification and cloning artefacts or from sequencing errors. In addition, the detection of a 48 nt and a 54 nt deletion in amplification products from cultivar H73-6110 might indicate the presence of two SCYLV genotypes in this plant line. RT-PCR with primers flanking this particular region yielded amplification products of the expected size; 409 bp from the resistant cultivar H87-4319 and about 359 bp from the susceptible cultivars H73-6110 and H87-4094 (Fig. 5b). When a few other cultivars were tested with the same primer combinations, H78-7750 also showed the deletion, whereas two cultivars obtained from Cuba, JA-605 and CP52-43 contained the 50 nt stretch (Fig. 5b). The deletion in SCYLV from susceptible cultivars lies in the ORF1 for a “multifunctional protein” which is thought to be involved in suppression of gene silencing, and at a cleavage point of RNA-dependent RNA polymerase (RdRp, ORF1 to ORF2). The amino acid sequences of RNA-dependent RNA polymerase (RdRp) from fully sequenced SCYLV-strains showed lower sequence identities in the first half and high identity in the second half of the protein (Fig. 6). The 16–18 aa gap of the two isolates H73-6110 and H87-4094 lies just in between of these two halves.

Sequence gap in SCYLV from susceptible cultivars (a) and RT-PCR of the sequence segment containing the deletion (b). a ORFs for coded proteins and location of the sequence gap in SCYLV from susceptible cultivars versus SCYLV from resistant cultivar. The gap was in overlap of fragments A and B. b RT-PCR of the sequence segment in ORF1 which contains the deletion in some cultivars. Primers YL1FOR and YL1REV were designed to amplify the sequence nt1211-1620 from RNA-preparations of sugarcane leaves as templates. Cultivars H73-6110 and H87-4094 are susceptible, cvs. H87-4319 and H78-7750 are resistant. JA-605 and CP52-43 are cultivars from Cuba. M: 1 kb and 50 bp DNA molecular size markers (Fermentas, St. Leon Rot, Germany). The lower panel shows the transcription of ribosomal RNA in the same preparation to demonstrate the activity of cDNA amplification (108 bp from 25S rRNA). The PCR products were electrophoresed on 1% agarose gel and stained with ethidium bromide

Alignment of the deduced amino acid sequences of RdRp of Hawaiian and some other SCYLV-strains. Asterisks indicate perfect matches with all sequences. Columns denote amino acid differences. The x denotes a place where the nucleotide identity was ambiguous
Discussion
All tested Hawaiian cultivars contained SCYLV, even the so-called resistant cultivars which previously had been thought to be virus-free based on TBIA (Schenck and Lehrer 2000). Obviously the immunological assay is less sensitive than RT-PCR even for the highly conserved YLS-segment. Only the cultivar line which had been generated by meristem tip culture was indeed virus-free. Semiquantitative RT-PCR indicated that the susceptible cultivars had a hundred-fold higher virus titre than the resistant cultivars. The clone of cultivar H65-7052 grown in the Bayreuth greenhouse had a very low titre which was unexpected because the same cultivar had been previously found to be moderately susceptible, i. e. containing SCYLV at a sufficient concentration to be detected by TBIA, although the level was strongly fluctuating (Lehrer et al. 2007). Either it happened that the RNA-extract from leaves was accidentally made when the fluctuating SCYLV-titre was at minimum or the particular clone in Bayreuth was a low-titre clone. Recent tests of H65-7052 in Hawaii seemed to indicate that plants of the cultivar H65-7052 with low SCYLV-titre “inherited” the low titre through vegetative seed pieces (Zhu et al. 2010), thus possibly differently proliferating virus strains coexist in this cultivar. We found in northern blots that RNA of Hawaiian SCYLV is divided into genomic RNA and two subgenomic RNAs (Fig. 2) with estimated sizes of 6.0, 2.4 and 1.0 kb, similar to 6.0, 2.4 and 0.8–1.0 kb reported previously (Borth et al. 1994; Moonan et al. 2000). Thus SCYLV may be similar to other plant RNA viruses, which have evolved numerous strategies of genome expression to invade host plants, for example, divided genomes, subgenomic messenger RNAs, frame shifting, overlapping reading frames or stop codon suppression (Zaccomer et al. 1995).
The fragments A–D of SCYLV from Hawaiian cultivars were amplified using the published primer sequences (Abu Ahmad et al. 2006). Not all fragments could be amplified at the same quantity and some fragments from some cultivars were not amplified at all. A similar result was reported by Abu Ahmad et al. (2006). The reasons are unknown but it may be due to sequence divergences in the primer binding regions. The fragment YLS was easily amplified in all SCYLV-preparations and it turned out to be the most conserved region. Three Hawaiian SCYLV-strains isolated from three different cultivars were fully sequenced in this study. We found that the Hawaiian strains (including 3 previously published SCYLV-fragments from Hawaiian cultivars, Haw1-3) are constantly grouped together and often located next to strains from Peru and Brazil. On the whole genome level the PER strains are next to Hawaii isolates and apart from the BRA strains (Fig. 4f). This close relationship may be explained by the fact that the Peru-strain was isolated from sugarcane cultivars, which were developed in Hawaii and exported to Peru, probably already infected with SCYLV in Hawaii. A SCYLV-genome isolated from a Florida cultivar grouped next to these virus strains. From the phylogenetic analysis we propose that a new SCYLV-group is defined namely a HAW-PER group, or, alternatively, as a subgroup of the BRA-strains, the next relatives to the PER and HAW strains. It would be interesting to analyse the SCYLV-strains from the same cultivars which are currently grown in the Hawaiian breeding station to see, whether the small differences to the Peru-strains were already present in Hawaii or whether they were derived from sequence changes or recombinations with BRA strains in the past 30 years in Peru. The Florida strain also clustered into the Haw-Per group, either because SCYLV in Florida is generally closely related to the Hawaiian SCYLV, or the analysed plant happened to become infected by the Hawaiian strain, because many SCYLV-infected Hawaiian cultivars had been imported to Florida in the late 1990s to replace cultivars which had been lost in a hurricane (Comstock et al. 2001). The REU strains and a recently published genome from a Chinese strain (CHN) represent distinct groups each (Fig. 4f).
The phylogenetic distances between SCYLV-strains are insignificant for the fragment YLS, which was already previously called a diagnostic sequence (Comstock et al. 1998). The coat protein encoded by fragment YLS is extremely conserved, only five amino acid differences were detected for all SCYLV-strains where CP deduced amino acid sequence is available. Therefore the immunological test for SCYLV by an antibody directed against the coat protein (Lockhart and Cronje 2000) is a valid diagnostic field test for the presence of the virus despite its limiting detection threshold. The CP is directly associated with the success of infection, as it is involved in viral transmission, particle packaging, and viral accumulation within the plant (Peiffer et al. 1997; Brault et al. 2003). Thus, a high degree of conservation in the CP protein sequence is expected.
In contrast, the other fragments exhibit phylogenetic distances up to twice as large as fragment YLS. The variations in RNA-sequence and deduced amino acid sequence were found to be relatively high in the RNA-dependent RNA-polymerase, as previously reported by Moonan and Mirkov (2002). There is a phylogenetic inconsistency between fragments B and C concerning ScYLV-C1 and –L1, which cluster together in fragment B, but are far apart in fragment C (Fig. 5c and d). Interestingly, the first half of fragment C was derived from Potato leaf roll virus, the second half from Barley yellow dwarf virus (Moonan et al. 2000). Possibly these two sequence parts may have diverged differently during evolution (or recombination) leading to an ambiguous result. The SCYLV-C1 sequence had been taken previously as evidence for the Colombian strain to belong to a progenitor population of the other SCYLV strains (Moonan and Mirkov 2002).
Sequence comparison of SCYLV between one resistant and two susceptible cultivars showed a 48 to 54 nt long deletion in SCYLV isolated from susceptible cultivars. This deletion is located in the RNA-dependent RNA-polymerase/silencing suppressor ORF1/2. The RNA-dependent RNA polymerase (RdRp) plays a central role in the replication of RNA viruses and it is tempting to speculate that theses deletions could play a role in controlling the proliferation rates of SCYLV, thereby increasing the SCYLV titre in the susceptible cultivars. Previously, short deletions of 3 and 1 nt in ORF2-3 have been reported in a Colombian strain and in cv. SP71-6163 (Moonan and Mirkov 2002) and a 25 nt deletion in fragment C related to ORF3 (Abu Ahmad et al. 2006). These reported gaps are obviously very different in size and position from that found in our case. A similar gap at the same position as in H73-6110 and H87-4094 was found in GenBank entry AJ491131 derived from cultivar CP65-357 (Smith et al. 2000), which is reported to be highly susceptible to YL (Lockhart and Cronje 2000). Amazingly, the nucleotide sequence of many GenBank entries start around the first nucleotides after the deletion, thus it is unknown whether the nucleotides before were absent or were eliminated. Future analysis of other susceptible and resistant cultivars should show whether the susceptibility for SCYLV can be correlated with the absence of the 48 to 54 nt stretches in ORF1/2. The reason for the deletion in the sequence of SCYLV from some cultivars is unknown, it could indicate the presence of two differently proliferating virus strains or a different splicing of the viral RNA by sugarcane cultivars. In the first case one would expect mixed SCYLV-infections of the cultivars, because the breeding station is highly infested by viruliferous aphids, which should result in a strong interchange of the two (or more) SCYLV-strains. In the second case the splicing of viral RNA by plant spliceosomes is essential so that the viral RNA has access to the nuclear space or to a cytosolic spliceosome (König et al. 2007). However, the flanking regions of the deleted sequence do not represent a general splicing signal. Future controlled infection experiments with viruliferous aphids may cast more light on SCYLV-susceptibility, whether it is a virus or a plant feature.
References
Abu Ahmad, Y., Rassably, L., Royer, M., Borg, Z., Braithwaite, K. S., Mirkov, T. E., et al. (2006). Yellow leaf of sugarcane is caused by at least three different genotypes of sugarcane yellow leaf virus, one of which predominates on the Island of Réunion. Archive of Virology, 151, 1355–1371.
Abu Ahmad, Y., Costet, L., Daugrois, J.-H., Nibouche, S., Letourmy, P., Girard, J.-C., et al. (2007). Variation in infection capacity and in virulence exists between genotypes of Sugarcane yellow leaf virus. Plant Disease, 91, 253–259.
Borth, W., Hu, J. S., & Schenk, S. (1994). Double-stranded RNA associated with sugarcane yellow leaf syndrome. Sugar Cane, 3, 5–8.
Brault, V., Bergdoll, M., Mutterer, J., Prasad, V., Pfeffer, S., Erdinger, M., et al. (2003). Effects of point mutations in the major capsid protein of beet western yellows virus on capsid formation, virus accumulation, and aphid transmission. Journal of Virology, 77, 3247–3256.
Comstock, J. C., Irvine, J. E., & Miller, J. D. (1994). Yellow leaf syndrome appears on the United States mainland. Sugar Journal, 56, 33–35.
Comstock, J. C., Irey, M. S., Lockhart, B. E. L., & Wang, Z. K. (1998). Incidence of yellow leaf syndrome in CP cultivars based on polymerase chain reaction and serological techniques. Sugar Cane, 4, 21–24.
Comstock, J. C., Miller, J. D., & Schnell, R. J. (2001). Incidence of Sugarcane yellow leaf virus in clones maintained in the World Collection of Sugarcane and Related Grasses at the United States National Repository in Miami, Florida. Sugar Technology, 3, 128–133.
Guindon, S., & Gascuel, O. (2003). A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Systematic Biology, 52, 696–704.
Komor, E., ElSayed, A., & Lehrer, A. T. (2010). Sugarcane yellow leaf virus introduction and spread in Hawaiian sugarcane industry: Retrospective epidemiological study of an unnoticed, mostly asymptomatic plant disease. European Journal of Plant Pathology, 127, 207–217.
König, H., Matter, N., Bader, R., Thiele, W., & Müller, F. (2007). Splicing segregation: the minor spliceosome acts outside the nucleus and controls cell proliferation. Cell, 131, 718–729.
Lehrer, A. T., & Komor, E. (2008). Symptom expression of yellow leaf disease in sugarcane cultivars with different degrees of infection by Sugarcane yellow leaf virus. Plant Pathology, 57, 178–89.
Lehrer, A. T., Schenck, S., Yan, S.-L., & Komor, E. (2007). Movement of aphid-transmitted sugarcane yellow leaf virus(ScYLV ) within and between sugarcane plants. Plant Pathology, 56, 711–717.
Lockhart, B. E. L., & Cronje, C. P. R. (2000). Yellow leaf syndrome. In P. Rott, R. A. Bailey, J. C. Comstock, B. J. Croft, & A. S. Saumtally (Eds.), A guide to sugarcane diseases (pp. 291–295). Montpellier: La Librairie du Cirad.
Moonan, F., & Mirkov, T. E. (2002). Analyses of genotypic diversity among North, South, and Central American isolates of Sugarcane yellow leaf virus: evidence for Colombian origins and for intraspecific spatial phylogenetic variation. Journal of Virology, 76, 1339–48.
Moonan, F., Molina, J., & Mirkov, T. E. (2000). Sugarcane yellow leaf virus: an emerging virus that has evolved by recombination between luteoviral and poleroviral ancestors. Virology, 269, 156–171.
Peiffer, M. L., Gildow, F. E., & Gray, S. M. (1997). Two distinct mechanisms regulate luteovirus transmission efficiency and specificity at the aphid salivary gland. The Journal of General Virology, 78, 495–503.
Sambrook, J., & Russell, D. W. (2001). Molecular cloning: a laboratory manual (3rd ed.). Cold Spring Harbor: Cold Spring Harbor Laboratory Press.
Schenck, S. (1990). Yellow leaf syndrome—a new sugarcane disease. Hawaiian Sugar Planters Association: Annual Report 38.
Schenck, S., & Lehrer, A. T. (2000). Factors affecting the transmission and spread of Sugarcane yellow leaf virus. Plant Disease, 84, 1085–1088.
Smith, G. R., Borg, Z., Lockhart, B. E. L., Braithwaite, K. S., & Gibbs, M. J. (2000). Sugarcane yellow leaf virus: a novel member of the Luteoviridae that probably arose by interspecies recombination. The Journal of General Virology, 81, 1865–1869.
Thompson, J. D., Higgins, D. G., & Gibson, T. J. (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Research, 22, 4673–4680.
Vega, J., Scagliusi, S. M. M., & Ulian, E. C. (1997). Sugarcane yellow leaf disease in Brazil: evidence of association with a Luteovirus. Plant Disease, 81, 21–26.
Zaccomer, B., Haenni, A.-L., & Macaya, G. (1995). The remarkable variety of plant RNA virus genomes. Journal of General Virology, 76, 231–147.
Zhu, Y. J., Lim, S. T. S., Schenck, S., Arcinas, A., & Komor, E. (2010). RT-PCR and quantitative real-time RT-PCR detection of Sugarcane Yellow Leaf Virus (SCYLV) in symptomatic and asymptomatic plants of Hawaiian sugarcane cultivars and the correlation of SCYLV titre to yield. European Journal of Plant Pathology, 127, 263–273.
Acknowledgement
Sugarcane cultivars have been given by Dr. A. Lehrer, HARC and by Dr. I. Medina Borges and Dr. E. Ortega, Habana. The help and advice by Dr. Trampczynska and S. Rentsch (Bayreuth, Germany) is gratefully acknowledged. A. Elsayed is grateful to the Egyptian Government for the stipendium. We also thank Bayreuth University for a special fund and Fonds der Chemischen Industrie.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
ElSayed, A.I., Weig, A.R. & Komor, E. Molecular characterization of Hawaiian Sugarcane yellow leaf virus genotypes and their phylogenetic relationship to strains from other sugarcane-growing countries. Eur J Plant Pathol 129, 399–412 (2011). https://doi.org/10.1007/s10658-010-9703-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10658-010-9703-0