
Abstract
Citrus Huanglongbing (HLB) is a devastating disease of citrus known to be associated with a fastidious, phloem-limited Gram-negative, yet to be cultured bacterium in the genus Candidatus Liberibacter. In the present study we have developed a method to quantify viable Candidatus Liberibacter asiaticus (Las) with the aid of ethidium monoazide (EMA) which can differentiate live from dead cells. First, calibration curves were developed with the aid of quantitative real-time PCR (QPCR) by using a plasmid template consisting of a 703 bp DNA fragment of rplKAJL-rpoBC (β-operon) region. Standard equations were then developed to quantify Las genome equivalents in citrus, periwinkle, and Asian citrus psyllid, Diaphorina citri. To overcome the limitation of quantitative PCR in discriminating between live and dead bacterial cells, EMA was used to inhibit the amplification of DNA from the dead cells of Las in plant samples. By using the standard equations and EMA-QPCR methods developed in this study, we found that the proportion of viable cells in citrus and periwinkle ranged from 17–31% and 16–28%, respectively. It was determined that a minimum bacterial concentration is required for HLB symptom development by quantifying the population of Las in symptomatic and asymptomatic leaves. The EMA-QPCR methodology developed in the present study should provide an accurate assessment of viable HLB pathogen, providing a tool to investigate disease epidemiology and thus act as a crucial component for disease assessment and management.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Citrus Huanglongbing (HLB) also called citrus greening is one of the most devastating diseases of citrus, causing severe losses and significantly affecting the world citrus industry (Halbert and Manjunath 2004). The disease is associated with a phloem-limited pathogenic bacterium which belongs to the genus Candidatus Liberibacter spp. (Jagoueix et al. 1994). Currently, three species of the pathogen have been identified: Candidatus Liberibacter asiaticus (Las), Candidatus Liberibacter africanus and Candidatus Liberibacter americanus (Lam), of which Las is more prevalent (Bové 2006). Las is naturally vectored in citrus by the Asian citrus psyllid, Diaphorina citri, and can be artificially transmitted by grafting with infected plant materials (Christensen et al. 2004; Dahllöf et al. 2000; Garnier and Bové 1983, 1993).
Characteristic symptoms of HLB include blotchy mottling with green islands on leaves. Other common symptoms include stunting, fruit decline, and small, lopsided fruits with poor colouration (Bové 2006). Previous studies have indicated that HLB infection causes disorder in the phloem and severely impairs assimilate translocation in host plants. The mechanism behind the phloem disorder is still unknown. It has been speculated that the HLB pathogen is present in the citrus phloem at low titer (Li et al. 2006). However, with the exception of some microscopic studies, not enough is understood regarding the precise bacterial concentration in planta (Bové 2006). Also, it is unknown if there is a relationship between Las concentration in planta and symptom development.
Quantitative assays are useful for determining the virulence mechanism(s) of pathogens, infection ability of insect vectors, and development of efficient management strategies (Alvarez 2004; Bach et al. 2002). Since the efforts to isolate Las in pure culture have been unsuccessful, accurate quantification remains difficult. Several methods that have been used to quantify Liberibacter species and other endophytic bacteria such as phytoplasma include competitive PCR, nested competitive PCR, and quantitative DNA hybridisation (Blomquist and Kirkpatrick 2002; Jarausch et al. 1998; Kawabe et al. 2006). However, these commonly used molecular methods for quantification have several limitations. Classical PCR assays are not quantitative and need separation of the products on agarose gels and visualisation under UV light (Salm and Geider 2004). DNA hybridisation methods are time-consuming and labour-intensive. Recently, quantitative real-time PCR based on the primers from 16S rDNA (Li et al. 2006; Teixeira et al. 2008) and rplKAJL-rpoBC (β-operon) (Hocquellet et al. 1997; Wang et al. 2006) have been used for detection and quantification of the HLB pathogen. However, these assays could not differentiate between viable and dead cells. Bacterial genomic DNA can remain stable for up to 3 weeks after cell death (Josephson et al. 1993). Consequently, the above described assays are likely to substantially overestimate the population of HLB pathogen in the hosts. Considering the severe threat posed by greening disease to the Florida citrus industry, there is a critical need to accurately quantify Las, which should improve disease management and understanding of the virulence mechanism(s) of Las.
Ethidium monoazide (EMA) PCR has been reported to effectively discriminate between live and dead cells (Nocker and Camper 2006; Rudi et al. 2005; Wang and Levin 2006). This diagnostic DNA-based method combines the use of a live-dead discriminating dye with the speed and sensitivity of quantitative PCR. Viable and dead discrimination is obtained by covalent binding of EMA to DNA of dead cells by photoactivation (Wagner et al. 2008). EMA penetrates only the dead cells with compromised membrane/cell systems. Subsequent photoinduced cross-linking inhibits the PCR amplification of DNA from dead cells (Soejima et al. 2007). Nocker and Camper (2006) have reported that in addition to inhibition of amplification, DNA yield from the dead cells is affected during the DNA extraction procedure. Although EMA has been used to facilitate the quantitative real-time PCR amplification of targeted DNA sequences of various human pathogens (Nocker and Camper 2006; Rudi et al. 2005; Wang and Levin 2006), there is just one report of using this technique to differentiate viable and dead cells of the plant pathogenic bacterium Clavibacter michiganensis subsp. michiganensis (Luo et al. 2008). No studies have yet been undertaken to apply EMA-QPCR for determining the exact number of viable cells of the HLB pathogen or any other uncultivable bacteria. Attempts were made in this study to quantify viable Las cells with the aid of EMA. The rapid and quantitative PCR methodology developed in the present study should provide an accurate assessment of the viable HLB pathogen, providing a tool to investigate disease epidemiology and thus act as a crucial component for disease assessment and management.
Materials and methods
PCR
All conventional PCR reactions were performed in a BIORAD DNAEngine® Peltier thermal cycler (Bio-Rad Hercules, CA, USA) using 2× PCR Master Mix (Promega, Madison, WI, USA) containing 50 units ml−1 of Taq DNA polymerase, 400 μM (each) dNTPs and 3 mM MgCl2. Amplification of the DNA was performed in 50 μl total volumes using 0.4 μM of each primer and 100 ng of DNA template. The PCR conditions were 95°C for 5 min followed by 35 cycles of 30 s of denaturation at 95°C, 30 s of annealing at 55°C, and 1 min of extension at 72°C.
Cloning and sequencing
The HLB-specific plasmid template used in this study, pLBA2, was developed by cloning a portion of DNA sequence of the Las rplKAJL-rpoBC (β-operon)region (Hocquellet et al. 1999). The resulting 703 bp fragment was amplified as described above using Liberibacter-specific primers A2 and J5 (Hocquellet et al. 1999) and DNA was extracted from infected leaf midribs as a template. The gene sequence for rplKAJL-rpoBC (β-operon) was obtained from GenBank (Accession no: AY342001.1). The PCR products were electrophoresed on 1% agarose gel and the expected size DNA band was excised and purified using Wizard® SV gel and PCR clean-up system (Promega). The purified DNA was ligated to pGEM T-easy vector (Promega) and transformed into chemically-competent Escherichia coli (DH5α). The positive clones were confirmed by restriction digestion analysis and sequencing of the resultant plasmid (pLBA2) (data not shown).
Plant and psyllid materials and extraction of DNA
Symptomatic leaf samples (fully expanded and hardened) were collected from Las-infected sweet orange trees (Citrus sinensis) (about 5 year-old), which were previously PCR-confirmed, from citrus groves in Polk County, Florida, USA. Asymptomatic leaf samples at a similar development stage were collected from healthy looking sweet orange trees (about 5 year-old) from the same citrus groves. In addition, branches (dark green and angular to slightly rounded in cross section), and root samples were collected from the same set of infected and healthy looking trees. Healthy and infected periwinkle (Catharanthus roseus) leaves as well as Asian citrus psyllids were obtained from biosecurity quarantine greenhouse facilities at the Citrus Research and Education Center, Lake Alfred, FL. For determining the development of Las in planta and the effect of age on Las concentration, leaf samples were collected in May, August, and November 2007 from sweet oranges maintained in the biosecurity greenhouse infected by grafting with infected buds and shoots in February 2007.
The plant material was washed with sterile distilled water (SDW). Midribs were separated from leaf. Midrib and root samples were cut into pieces and 0.1 g (fresh weight) of tissue from each sample was frozen in liquid nitrogen and stored at −80°C. Psyllids were stored in 70% ethanol until DNA extraction. DNA from plant samples was extracted using the Wizard® Genomic DNA purification kit (Promega) following the protocol for isolating genomic DNA from plant tissue. The DNA pellet was dried in Vacufuge (Eppendorf, Westbury, NY) for 15 min and dissolved in 100 μl of DNA rehydration solution (Promega). For DNA extraction from psyllids, each adult psyllid was homogenised in 300 μl TEN extraction buffer [STE buffer (10 mM Tris-Hcl pH 8.0, 1 mM EDTA pH 8.0 and 0.1 M NaCl) and 2% SDS] and 30 μl of proteinase-K (Qiagen) in a 1.5 ml eppendorf tube using a sterile plastic plunger. The homogenate was incubated at 50°C for 2–3 h. The samples were brought to room temperature and 400 μl of phenol:chloroform:isoamyl alcohol (24:24:1, vol/vol/vol) was added and subsequently centrifuged for 10 min at 18,000 g. The supernatant was treated with chloroform:isoamyl alcohol (24:1, vol/vol) followed by centrifugation for 10 min at 18,000 g. The supernatant (200 μl) was carefully collected in a clean tube and DNA was precipitated by adding 20 μl of 3 M sodium acetate (pH 5.2) and 1 ml of ice cold ethanol. At this point the tubes were inverted several times and placed at −80°C for 30 min. The precipitated nucleic acids were pelleted by centrifugation for 7 min at 18,000 g. The pellet was washed once with 1 ml of 70% ethanol, dried in Vacufuge for 15 min and dissolved in 50 μl of TE buffer.
Preparation of plasmid DNA and determination of standard curve
Bacterial plasmid DNA pLBA2 was isolated from an overnight culture using a Wizard® miniprep DNA purification system (Promega). The concentration and purity of DNA were determined using spectrophotometer ND-1000 (NanoDrop Technologies, Wilmington, DE, USA). The number of plasmid copies was calculated based on molecular weight using the formula: \({\text{Number}}\;{\text{of}}\;{\text{copies}} = {{\left( {{\text{Amount}}\;{\text{in}}\;{\text{ng}} \times {\text{Avogadro}}\prime {\text{s}}\;{\text{number}}} \right)}} \mathord{\left/ {\vphantom {{{\left( {{\text{Amount}}\;{\text{in}}\;{\text{ng}} \times {\text{Avogadro}}\prime {\text{s}}\;{\text{number}}} \right)}} {{\left( {{\text{Length}}\;{\text{in}}\;{\text{bp}} \times 1 \times 10^{9} \times 650} \right)}}}} \right. \kern-\nulldelimiterspace} {{\left( {{\text{Length}}\;{\text{in}}\;{\text{bp}} \times 1 \times 10^{9} \times 650} \right)}}\). The average weight of a base pair is assumed to be 650 Da and Avogadro’s number is 6.022 × 1023. Purified plasmid DNA was stored at −20°C until further use. To identify the detection limit and to develop a standard curve, a known concentration of plasmid in a series of dilutions ranging from 2 × 106 to 2 × 100 μl−1 was used. The reaction mixture without the plasmid DNA was used as a negative control.
Quantitative real-time PCR (QPCR)
All QPCR assays were performed in a 96-well plate using an ABI PRISM 7500 Sequence detection system (Applied Biosystems, Foster City, CA, USA). Primer/probe combinations, CQULA04F-CQULAP10-CQULA04R, were used to target the β-operon region of Las. The specificity of primer/probe CQULA04F-CQULAP10-CQULA04R has been confirmed previously (Wang et al. 2006). The probes were labelled with 56-FAM as a reporter fluorescent dye at the 5′ end and with 3’BHQ_1 as the quencher dye. QPCR reactions were performed according to the conditions described previously with slight modifications (Wang et al. 2006). Briefly, QPCR reactions were performed in a 25 μl reaction using a 2× Quantitect Probe PCR master mix (Qiagen, Valencia, CA, USA), 0.8 μM of each primer, 0.4 μM of probe (IDT, Coralville, IA, USA) and an appropriate amount of template DNA for generation of the standard curve. One μl of DNA was used as a template to quantify Las for the plant tissue samples and for the psyllid samples. The average Ct values used for estimating Las were determined by using the equation generated from the log curve. Later, the Las concentration was normalised to μg of total DNA. The PCR conditions were 50°C for 2 min, 95°C for 15 min, 45 cycles of 94°C for 15 s and 60°C for 1 min. Each individual sample was replicated 4 times on a 96-well plate and the whole reaction is repeated twice to verify the consistency of the method. Results were analysed using ABI Prism software. Raw data were analysed using the default settings (threshold = 0.2) of the software.
EMA pretreatment
The bark tissue of the infected citrus and periwinkle was collected by scraping the inner bark of branches with a sterile razor blade. The scrapings were collected, pooled together and diced into small pieces (about 1 mm or less) with the razor blade and an appropriate amount of water (to keep the scrapings submerged) was added. The samples were vortexed for 5 min, transferred into 50 ml conical tubes, and centrifuged at 1,200 g for 2 min. The supernatant was aliquoted and centrifuged at 12,000 g for 5 min. The supernatant was discarded and the pellet was used for EMA pretreatment. The leaf and root samples were procured as described above. The method of EMA pretreatment was adopted from Nocker and Camper (2006) and modified partially as below. EMA (Molecular Probes, Invitrogen, Carlsbad, CA, USA) was dissolved in water (in the dark) to yield a stock solution of 5 mg ml−1 and stored at −20°C. Approximately 40 mg of plant tissue was placed in 1.5 ml microcentrifuge tubes. One ml of SDW was added and the tubes were vortexed for 30 s to properly mix the sample. EMA was added (in the dark) from the stock solution to the sample tubes to a final concentration of 100 μg ml−1. The tubes were maintained in the dark at room temperature for 5 min with occasional flipping to allow the EMA to penetrate dead cells with compromised cell walls and to bind to their DNA. To activate and photolyse the EMA, the sample tubes with their lids off were exposed to light for 1 min from a halogen bulb (650 W) placed at a distance of 15 cm. The tubes were placed on ice during the light exposure to avoid excessive heating. Samples were then centrifuged at 5,000 g for 5 min and the supernatant was discarded. The pellet was then used for DNA extraction and subsequently QPCR by the methods described in previous sections. Samples that did not receive EMA were treated as a control. We used an equal volume (1 μl) of genomic DNA preparations from EMA-treated and control samples for quantitative PCR. A total of 15 samples from each tissue type were analysed to quantify viable versus dead Las cells. EMA was not used to distinguish between live and dead cells in psyllids due to the select agent status of Las in the USA and associated strict regulations with handling infected psyllids.
Quantification of Las in samples
Quantification of Las in plant and psyllid samples was done by QPCR, using the derived standard equations. To account for the effect of variations in the total amount of DNA yield from the same set of starting material, we have represented the predicted bacterial concentration as μg−1 of the total DNA wherever appropriate. The bacterial concentrations have also been presented as g−1 of fresh tissue without normalisation. The Ct values of unknown samples >32, which is the reliable detectable limit of Las concentration corresponding to 2 × 102 copies based on the logarithmic standard curve, were not used for quantification.
To test the effect of plant and psyllid DNA extracts on QPCR, assays similar to that described above were conducted using pLBA2, but in the presence of 50 ng of DNA (per reaction) extracted from healthy citrus leaf midribs or roots or psyllids. Other QPCR conditions such as the reaction volume, concentration of primers/probe and template were not altered. DNA samples of healthy citrus obtained from a greenhouse and psyllids fed on healthy citrus in a greenhouse were included as negative controls.
EMA was not used for all the samples due to the large number of samples analysed in this study. An estimate of the viable Las cells was calculated based on the ratio of viable to dead cells in the citrus leaves calculated using a subset of 15 samples for the 100 total samples as described above.
Results
Development of standard curve
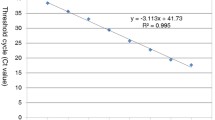
By using pLBA2 as a template, the plasmid concentration of 2 × 102 gave a consistent fluorescent signal with an average Ct value of 32 and hence was defined as the detection limit for the conditions used in this study. The lowest plasmid concentrations of 2 × 101 and 2 × 100 μl−1 did not amplify consistently and hence were not used in further analysis. The negative control did not have any detectable fluorescence above the threshold value. The logarithmic standard curve developed using five dilutions (2 × 106 to 2 × 102) of plasmid showed a very good fit for pLBA2 (Fig. 1). The R 2 value of 0.9994 indicated a high accuracy over a wide range of concentrations. No significant differences (P > 0.05) were observed between individual runs of the same assay indicating high sensitivity and reproducibility of the method (data not shown).

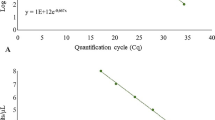
Effect of DNA extracts from citrus midrib, roots, and psyllids in QPCR assays. Each data point is the mean of eight replicates from two individual QPCR assays. Error bars indicate standard deviation
Effect of plant and psyllid DNA extracts on QPCR assays
As shown in Fig. 1, in comparison to plasmid DNA, leaf midrib, root or psyllid DNA extracts had no or a modest inhibitory effect on the amplification of the target DNA. To negate any unidentified inhibitory effect, separate standard curves were optimised using the Ct values obtained in the presence of leaf midrib or root or psyllid DNA extracts. Standard equations were generated by plotting the mean Ct values against the natural log concentrations of the plasmid (not shown) to predict the approximate concentration of Las genome equivalents. The equations were \(Y = - 0.288 \times \left( {{\text{Ct}}} \right) + 11.607\), \(Y = - 0.2768 \times \left( {{\text{Ct}}} \right) + 11.677\) and \(Y = - 0.2818 \times \left( {{\text{Ct}}} \right) + 11.845\) for plasmid+citrus midrib, plasmid+citrus root and plasmid+psyllid DNA, respectively.
Quantification of Las in citrus, periwinkle and psyllid
Citrus leaf midrib and root samples tested earlier for the presence of Las using traditional PCR (data not shown) were used to quantify the approximate concentration of bacteria. The Las concentration in the symptomatic leaf midrib and roots of sweet orange ranged from 3.43 × 105 to 1.37 × 106 and 6.67 × 103 to 9.39 × 105genome equivalents μg−1 of the total DNA, respectively (Table 1).
We also tested whether the method could be used to quantify Las from periwinkle leaves and citrus psyllids. Infected and uninfected (control) periwinkle and psyllids were used for this study. Las was transmitted to periwinkle via dodder (Garnier and Bové 1983) and to psyllids by feeding on infected citrus plants under quarantine greenhouse conditions. Prior to QPCR quantification, the presence of Las in the samples was tested by conventional PCR (data not shown). The data indicated that the concentration of Las ranged from 1.37 × 106 to 1.66 × 107 genome equivalents μg−1 of the total DNA in periwinkle midribs and laminae and 1.68 × 105 to 2.14 × 107 genome equivalents μg−1 of the total DNA in psyllids, respectively (Table 1).
Differentiation of viable from dead cells using EMA-QPCR
This QPCR-based quantification method targets the genome of the HLB pathogen through amplification of the β-operon region and could not differentiate between living and dead cells. We therefore used the method along with EMA pretreatment to quantify the number of viable Las cells in citrus leaves and periwinkle, the results are presented in Fig. 2a, b, respectively. The amplification of target DNA derived from dead cells in QPCR was inhibited when EMA was used. A significant reduction in the genome equivalents μg−1 of total DNA was observed when EMA was used in quantification studies. In the case of citrus, the number of Las cells was approximately 69%, 72% and 83% lower in EMA-treated leaf midrib, bark and root samples, respectively compared with the control. Similarly, a reduction of approximately 72% in root and leaf midrib and 84% in bark was observed in EMA-treated periwinkle samples compared with the control. The results suggested that a major proportion of Las was present in a non-viable state in the plant tissue.

Quantification of Las (genome equivalents μg−1 of total DNA) in EMA-treated and non-treated plant tissue of sweet orange (a) and periwinkle (b). Each bar represents the mean of three independent experiments each containing 15 replications
Minimal Las concentration required for HLB symptoms
The method developed in this study was applied to determine the possible correlation between appearance of HLB symptoms on leaves and Las concentration in the leaves. To test this hypothesis, only fully expanded leaves at a similar development stage with or without blotchy mottle symptoms were collected from citrus groves. QPCR analysis was performed and the approximate concentrations of Las were predicted using the equation developed for the midrib. In total, 100 asymptomatic samples and 100 symptomatic samples were tested with QPCR. An estimate of the viable Las cells was calculated based on the ratio of viable to dead cells, calculated using a subset of 15 samples from the 100 total samples. Interestingly, a considerably large concentration of Las was observed in seven of 100 asymptomatic leaves(Table 2). However, the mean Las concentration from asymptomatic leaves was significantly lower (P < 0.05) than the mean Las concentrations from all symptomatic leaves (Table 2). All of the symptomatic leaves exhibited a higher concentration of the HLB bacterium ranging from 9.17 × 105 to 6.60 × 106 genome equivalents μg−1 plant DNA or approximately 2.84 × 105 to 2.05 × 106 genome equivalents of viable Las μg−1 plant DNA. Seven asymptomatic leaves had a concentration ranging from 7.84 × 103 to 4.81 × 105 genome equivalents μg−1 plant DNA or 2.43 × 103 to 1.49 × 105 genome equivalents of viable Las μg−1 plant DNA while Las was not detected in the remaining non-symptomatic leaves. These results indicate a specific Las threshold concentration for symptom development.
Dynamic development of Las concentration in planta
QPCR analysis was performed to monitor the concentration of Las over time after infection in planta. All tested plants clearly showed a gradual increase in Las concentration from the time of infection. The mean Las concentration for May 2007 (1.8 × 105 μg−1 of total DNA) was significantly (P < 0.05) lower than August 2007 (1.77 × 106 genome equivalents μg−1 of total DNA). To further understand its distribution in young and old tissues, leaves were collected from three different points (representing young to mature tissues) on the infected shoot. The data revealed that the mean Las concentrations in old (9.62 × 105), mid-age (2.09 × 106) and young (1.77 × 106) leaves of the shoot were not significantly (P < 0.05) different from each other.
Discussion
QPCR enables quantification of nucleic acids in unknown samples by a direct comparison with standards amplified in parallel reactions (Morrison et al. 1999). In this study, we have successfully developed a method for quantification of live Las cells in citrus and periwinkle using QPCR assays with the aid of EMA. The standard equations for the quantification of Las were developed using a Las-specific artificial template consisting of a 703 bp β-operon fragment. All sequenced bacteria contain one copy of the β-operon region (Dahllöf et al. 2000). It is very likely that Las also contains one copy of the β-operon region which in this study corresponds to one bacterium. Thus quantification of the copy numbers of the β-operon region should allow calculation of the Las population without culturing the causal bacterium. The 16S rDNA was not used to develop the standard curve due to the usual varied copy numbers in the genomes of bacteria (Rainey et al. 1996). Similar methods were used earlier for accurate quantification of phytoplasma (Aldaghi et al. 2006) and virus (Renz et al. 2006). In comparison with other similar developed procedures, this method was optimised to take into account the potential effect of inhibitors and can be used to accurately quantify Las from citrus, periwinkle and insect tissues. In addition, the method was applied to study the relationship between appearance of HLB symptoms and Las concentration.
Plant samples often contain organic substances such as polyphenolic and polysaccharides with the potential to inhibit PCR assays (Aldaghi et al. 2006; Lin et al. 2006; Zhang and Lin 2005). Thus, the effective removal of PCR inhibitors from samples has to be considered for any survey using quantitative real-time PCR assays. This can be done by selecting a DNA purification procedure and DNA polymerase from a wide range of commercially available kits that suit the purpose. The inhibitory effects of contamination could be reversed by inclusion of polyvinylpyrrolidone in the PCR (Koonjul et al. 1999). Aldaghi et al. (2006) have reported overcoming PCR inhibition by diluting the DNA extracts that minimised the inhibitory effects of PCR. We have also observed a similar inhibitory effect due to the presence of DNA extracts on conventional PCR (data not shown). In order to overcome the inhibitory effect, the equations generated in this study were optimised by taking into consideration the effect (if any) of potential plant or psyllid inhibitors.
The inability to differentiate between DNA from viable and dead bacterial cells is a major obstacle in DNA-based molecular diagnostics. These quantification methods significantly extrapolate the number of viable cells in mixed populations (Nocker and Camper 2006). Other methods for determination of viable cells such as those utilising RNA-based diagnostics are expensive and time-consuming (Alvarez 2004; Norton and Batt 1999). EMA allows selective amplification of DNA by QPCR from only viable and not dead cells, which substantially increases the utility of PCR for rapidly determining and quantifying the presence of targeted viable microorganisms in environmental samples without enrichment (Rudi et al. 2005). The discrimination of live versus dead cells occurs both during DNA extraction and PCR amplification (Nocker and Camper 2006). Our results are in accordance with earlier reports which have shown that quantitative analysis of total DNA can lead to a substantial overestimation of the presence of living microorganisms and the accompanying pathogenic threats (Luo et al. 2008; Nocker and Camper 2006, Wang and Levin 2006). The use of EMA-PCR with the method developed in this study will enhance our ability to quantify Las accurately in plant samples and should increase our understanding of the biology of the HLB pathogen.
Using the method described in the present study, we have successfully quantified Las from citrus leaf midribs, roots, infected periwinkle and citrus psyllids. In our QPCR assays we observed that the bacterial concentration was consistently higher in artificially-infected periwinkle than field-infected citrus and also in experimentally-infected psyllids compared to natural psyllids collected from the field (data not shown). The higher titer of Las in periwinkle than in citrus is consistent with previous observations using electron microscopy (Bové 2006). Although the Las concentration varied widely across samples, this method can be used to accurately determine the concentration of Las from plants and psyllids. Also the consistency of results suggests that the method enables very specific, high-throughput, quantitative detection of the HLB pathogen over a wide range of hosts and should be easily adaptable for diagnostic purposes.
It was determined that a minimal Las concentration is required for HLB symptom development. All symptomatic leaves contained a higher concentration of HLB bacterium than asymptomatic leaves. This confirms the observations made using electron microscopic studies of symptomatic and asymptomatic midribs by Bové (2006). Higher Lam populations have also been reported in symptomatic leaves in Brazil (Teixeira et al. 2008). The fact that only seven of the 100 asymptomatic samples tested positive for Las may be due to the low concentration of Las, which may have escaped the detection limit. This strongly suggests that the population of Las should reach a threshold concentration prior to symptom development. A similar population threshold has been reported for disease symptoms caused by another endophytic plant pathogenic bacterium Xylella fastidiosa in grapevines and citrus (Hill and Purcell 1997; Li et al. 2003). The lower bacterial concentration and frequency of Las detected in asymptomatic leaves also explains the difficulty in detecting the HLB bacterium in asymptomatic leaves.
The actual Las concentrations in younger leaves were higher than in mature (lower) leaves, but the differences were not statistically (P < 0.05) significant. HLB bacterial concentration was higher in leaves than in roots. Previous reports have also indicated an uneven distribution of Las cells in different tissues of infected sweet orange trees (Tatineni et al. 2008). The present observation is also consistent with the findings for phytoplasma (Christensen et al. 2004). The concentration of HLB bacterium in citrus leaves was similar to the concentration of phytoplasma in the petioles (Aldaghi et al. 2006; Christensen et al. 2004). This may suggest that both endophytic bacteria are subjected to similar limitations in the phloem environment. In the present study we have also observed differences in the bacterial concentration after different sampling intervals following infection. These results suggested that the HLB bacterium grows very slowly in citrus. The slow growth rate may partially explain the extreme difficulty in culturing the HLB causal pathogen.
In the present study we have developed a method for quantification of viable Las using QPCR assays in different hosts including citrus and periwinkle with the aid of EMA. The molecular-based assay should prove useful for the pre-symptomatic diagnosis of HLB disease, monitoring and identification of live Las, epidemic studies, determination of virulence mechanism(s), and in the management of HLB.
References
Aldaghi, M., Massart, S., Roussel, S., & Jijakli, M. H. (2006). Establishment of a new method for a rapid and precise estimation of apple proliferation phytoplasma concentration in periwinkle. Communications in Agricultural and Applied Biological Sciences, 71, 853–857.
Alvarez, A. M. (2004). Integrated approaches for detection of plant pathogenic bacteria and diagnosis of bacterial diseases. Annual Review of Phytopathology, 42, 339–366. doi:10.1146/annurev.phyto.42.040803.140329.
Bach, H.-J., Tomanova, J., Schloter, M., & Munch, J. C. (2002). Enumeration of total bacteria and bacteria with genes for proteolytic activity in pure cultures and in environmental samples by quantitative PCR mediated amplification. Journal of Microbiological Methods, 49, 235–245. doi:10.1016/S0167-7012(01)00370-0.
Blomquist, C. L., & Kirkpatrick, B. C. (2002). Identification of phytoplasma strains and insect vectors of peach yellow leaf roll disease in California. Plant Disease, 86, 759–763. doi:10.1094/PDIS.2002.86.7.759.
Bové, J. M. (2006). Huanglongbing: A destructive, newly emerging, century-old disease of citrus. Journal of Plant Pathology, 88, 7–37.
Christensen, N. M., Nicolaisen, M., Hansen, M., & Schulz, A. (2004). Distribution of phytoplasmas in infected plants as revealed by real-time PCR and bioimaging. Molecular Plant–Microbe Interactions, 17, 1175–1184. doi:10.1094/MPMI.2004.17.11.1175.
Dahllöf, I., Baillie, H., & Kjelleberg, S. (2000). rpoB-based microbial community analysis avoids limitations inherent in 16S rRNA gene intraspecies heterogeneity. Applied and Environmental Microbiology, 66, 3376–3380. doi:10.1128/AEM.66.8.3376-3380.2000.
Garnier, M., & Bové, J. M. (1983). Transmission of the organism associated with citrus greening disease from sweet orange to periwinkle by dodder. Phytopathology, 73, 1358–1363. doi:10.1094/Phyto-73-1358.
Garnier, M., & Bové, J. M. (1993). Citrus greening disease and the greening bacterium. In P. Moreno, J. V. daGraca, & L. W. Timmer (Eds.), Proceedings of 12th conference of international organization of citrus virologists (pp. 212–219). Riverside, CA: IOCV.
Halbert, S. E., & Manjunath, K. L. (2004). Asian citrus psyllids (Sternorryhncha: Psyllidae) and greening disease of citrus: A literature review and assessment of risk in Florida. The Florida Entomologist, 87, 330–352. doi:10.1653/0015-4040(2004)087[0330:ACPSPA]2.0.CO;2.
Hill, B. L., & Purcell, A. H. (1997). Populations of Xylella fastidiosa in plants required for transmission by an efficient vector. Phytopathology, 87, 1197–1201. doi:10.1094/PHYTO.1997.87.12.1197.
Hocquellet, A., Bové, J. M., & Garnier, M. (1997). Production and evaluation of non-radioactive probes for the detection of the two Candidatus Liberobacter species associated with citrus huanglongbing (Greening). Molecular and Cellular Probes, 11, 433–438. doi:10.1006/mcpr.1997.0140.
Hocquellet, A., Toorawa, P., Bové, J. M., & Garnier, M. (1999). Detection and identification of the two Candidatus Liberobacter species associated with citrus huanglongbing by PCR amplification of ribosomal protein genes of the β operon. Molecular and Cellular Probes, 13, 373–379. doi:10.1006/mcpr.1999.0263.
Jagoueix, S., Bové, J. M., & Garnier, M. (1994). The phloem-limited bacterium of greening disease of citrus is a member of alpha subdivision of the Proteobacteria. International Journal of Systematic Bacteriology, 44, 379–386.
Jarausch, W., Lansac, M., Saillard, C., Broquaire, J. M., & Dosba, F. (1998). PCR assay for specific detection of European stone fruit yellows phytoplasmas and its use for epidemiological studies in France. European Journal of Plant Pathology, 104, 17–27. doi:10.1023/A:1008600828144.
Josephson, K. L., Gerba, C. P., & Pepper, I. L. (1993). Polymerase chain reaction detection of nonviable bacterial pathogens. Applied and Environmental Microbiology, 59, 3513–3515.
Kawabe, K., Truc, N. T. N., Lan, B. T. N., Hong, L. T. T., & Onuki, M. (2006). Quantification of DNA of citrus huanglongbing pathogen in diseased leaves using competitive PCR. Journal of General Plant Pathology, 72, 355–359. doi:10.1007/s10327-006-0306-8.
Koonjul, P. K., Brandt, W. F., Farrant, J. M., & Lindsey, G. G. (1999). Inclusion of polyvinylpyrrolidone in the polymerase chain reaction reverses the inhibitory effects of polyphenolic contamination of RNA. Nucleic Acids Research, 27, 915–916. doi:10.1093/nar/27.3.915.
Li, W. B., Pria Jr, W. D., Lacava, P. M., Qin, X., & Hartung, J. S. (2003). Presence of Xylella fastidiosa in sweet orange fruit and seeds and its transmission to seedlings. Phytopathology, 93, 953–958. doi:10.1094/PHYTO.2003.93.8.953.
Li, W. B., Hartung, J. S., & Levy, L. (2006). Quantitative real-time PCR for detection and identification of Candidatus Liberibacter species associated with citrus huanglongbing. Journal of Microbiological Methods, 66, 104–115. doi:10.1016/j.mimet.2005.10.018.
Lin, S., Zhang, H., & Dubois, A. (2006). Wide distribution and low abundance of Pfiesteria piscicida as detected by mtDNA-18S rDNA real-time PCR. Journal of Plankton Research, 28, 667–681. doi:10.1093/plankt/fbi150.
Luo, L. X., Wllters, C., Bolkan, H., Liu, X. L., & Li, J. Q. (2008). Quantification of viable cells of Clavibacter michiganensis subsp. michiganensis using a DNA binding dye and a real time PCR assay. Plant Pathology, 57, 332–337. doi:10.1111/j.1365-3059.2007.01736.x.
Morrison, T. B., Weiss, J. J., & Wittwer, C. T. (1999). Quantification of low-copy transcripts by continuous SYBR Green I monitoring during amplification. BioTechniques, 24, 954–962.
Nocker, A., & Camper, A. K. (2006). Selective removal of DNA from dead cells of mixed bacterial communities by use of ethidium monoazide. Applied and Environmental Microbiology, 72, 1997–2004. doi:10.1128/AEM.72.3.1997-2004.2006.
Norton, D. M., & Batt, C. A. (1999). Detection of viable Listeria monocytogenes with a 5′ nuclease PCR assay. Applied and Environmental Microbiology, 65, 2122–2127.
Rainey, F. A., Ward-Rainey, N. L., Janssen, P. H., & Hippe, H. (1996). Clostridium paradoxum DSM 7308(T) contains multiple 16S rRNA genes with heterogeneous intervening sequences. Microbiology, 142, 2087–2095.
Renz, K. G., Islam, A., Cheetham, B. F., & Walkden-Brown, S. W. (2006). Absolute quantification using real-time polymerase chain reaction of Marek’s disease virus serotype 2 in field dust samples, feather tips and spleens. Journal of Virological Methods, 135, 186–191. doi:10.1016/j.jviromet.2006.03.017.
Rudi, K., Moen, B., Drømtrop, S. M., & Holck, A. L. (2005). Use of ethidium monoazide and PCR in combination for quantification of viable and dead cells in complex samples. Applied and Environmental Microbiology, 71, 1018–1024. doi:10.1128/AEM.71.2.1018-1024.2005.
Salm, H., & Geider, K. (2004). Real-time PCR for detection and quantification of Erwinia amylovora, the causal agent of fireblight. Plant Pathology, 53, 602–610. doi:10.1111/j.1365-3059.2004.01066.x.
SAS Institute (2004). The SAS system for Windows. Release 9.1. Cary, NC: SAS Institute.
Soejima, T., Lida, K. I., Qin, T., Taniai, H., Seki, M., Takade, A., et al. (2007). Photoactivated ethidium monoazide directly cleaves bacterial DNA and is applied to PCR for discrimination of live and dead bacteria. Microbiology and Immunology, 51, 763–775.
Tatineni, S., Sagaram, U. S., Gowda, S., Robertson, C. J., Dawson, W. O., Iwanami, T., et al. (2008). In Planta distribution of ‘Candidatus Liberibacter asiaticus’ as revealed by polymerase chain reaction (PCR) and real-time PCR. Phytopathology, 98, 592–599. doi:10.1094/PHYTO-98-5-0592.
Teixeira, D. C., Saillard, C., Couture, C., Martins, E. C., Wulff, N. A., Eveillard-Jagoueix, S., et al. (2008). Distribution and quantification of Candidatus Liberibacter americanus agent of huanglongbing disease of citrus in Sao Paulo State, Brasil, in leaves of an affected sweet orange tree as determined by PCR. Molecular and Cellular Probes, 22, 139–150. doi:10.1016/j.mcp.2007.12.006.
Wagner, A. O., Malin, C., Knapp, B. A., & Illmer, P. (2008). Removal of free extracellular DNA from environmental samples by ethidium monoazide and propidium monoazide. Applied and Environmental Microbiology, 74, 2537–2539. doi:10.1128/AEM.02288-07.
Wang, S., & Levin, R. E. (2006). Discrimination of viable Vibrio vulnificus cells from dead cells in real-time PCR. Journal of Microbiological Methods, 64, 1–8. doi:10.1016/j.mimet.2005.04.023.
Wang, Z., Yin, Y., Hu, H., Yuan, Q., Peng, G., & Xia, Y. (2006). Development and application of molecular-based diagnosis for ‘Candidatus Liberibacter asiaticus’, the causal pathogen of citrus huanglongbing. Plant Pathology, 55, 630–638. doi:10.1111/j.1365-3059.2006.01438.x.
Zhang, H., & Lin, S. (2005). Development of a cob-18S rRNA gene real-time PCR assay for quantifying Pfiesteria shumwayae in the natural environment. Applied and Environmental Microbiology, 71, 7053–7063. doi:10.1128/AEM.71.11.7053-7063.2005.
Acknowledgements
We thank Dr. Madhulika Sagaram for her assistance with statistics. We thank Dr. Satyanarayana Tatineni and Dr. Chunxian Chen for critical review of this manuscript. This work has been supported by Florida Citrus Production Research Advisory Council (FCPRAC).
Author information
Authors and Affiliations
Corresponding author
Additional information
The authors P. Trivedi and U. S. Sagaram contributed equally to this work.
Rights and permissions
About this article
Cite this article
Trivedi, P., Sagaram, U.S., Kim, JS. et al. Quantification of viable Candidatus Liberibacter asiaticus in hosts using quantitative PCR with the aid of ethidium monoazide (EMA). Eur J Plant Pathol 124, 553–563 (2009). https://doi.org/10.1007/s10658-009-9439-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10658-009-9439-x