
Abstract
A monoclonal antibody that recognises components of the wall of sporangia of Peronospora destructor was raised. Tests using spores of higher fungi and other species of mildew demonstrated the specificity of the monoclonal. The antibody was used to develop lateral flow devices for sporangia of P. destructor. A competitive lateral flow format was developed which could detect onion downy mildew sporangia. Five-microliter gold anti-mouse IgM solution pre-mixed with 10 μl of P. destructor monoclonal antibody (EMA 242) proved the optimal concentration for detection of sporangia of P. destructor when applied to sample pads of lateral flow devices. Limits of approximately 500 sporangia of P. destructor could be detected by the absence of a test line on the lateral flow device within test samples. Using a scanning densitometer improved the sensitivity of detection. Further development and validation of the test is required if it is to be used for risk assessments of onion downy mildew in the field.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Foliar diseases of onion crops (onion downy mildew and Botrytis leaf blight) can cause heavy yield losses in bulb and salad onion crops. Onion downy mildew (Peronospora destructor) is the most serious disease in bulb and salad onions in the UK (Gilles et al. 2004; Clarkson et al. 2000). Actual yield losses in bulb onions of 60 to 75% have been recorded (Cook 1932; Cruickshank 1958). These losses mainly result from severe infections in bulb onion crops causing early defoliation, reduced bulb sizes and poor storage quality of bulbs (Rondomanski 1967). In salad onions, yield losses can be as high as 100% with whole crops being discarded, as downy mildew symptoms on the plant make them unmarketable. Fungicidal control of onion downy mildew is difficult and fungicides are only effective if they are applied before or immediately after disease first appears in the crop (Kennedy 1998). The environmental requirements for infection and sporulation by P. destructor have been reported (Yarwood 1937, 1943; Hildebrand and Sutton 1982). Mathematical models describing climatic effects on sporulation and infection have been described (Gilles et al. 2004; Battilani et al. 1996; Jesperson and Sutton 1987). However, despite the rapid development of onion downy mildew and the requirements for reductions in fungicide usage by consumers, the practical use of these systems in risk assessment has been limited to date.
New approaches in forecasting diseases of onion crops based on estimation of spore numbers in air samples have been reported (Carisse et al. 2005; Berger 1970). Detection and quantification of airborne spore numbers can be used to predict disease accurately before it is visible in the crop. Peaks of airborne spores are always detected prior to crops becoming infected. It has been reported that one or two peaks in sporangial concentration in the air of the potato blight pathogen Phytophthora infestans preceded the first observed symptoms of the disease in the field (Bugiani et al. 1998; Phillon 2003). In these studies the information on spore number had to be collected manually using a microscope which was slow and time consuming. Tests which can be conducted in the field are necessary if information on air-borne inoculum concentration is to be of more practical value. However, there are few reported systems for detecting and differentiating airborne spores (Wakeham et al. 2004). Molecular techniques exist for detection of spores using Hirst types spore samplers (Williams et al. 2001). Day et al. (2002) reported the development of cell flow cytometric differentiation of air-borne sporangia of P. infestans using ‘in field’ systems. Both techniques required either laboratory processing of results or the development and use of sensitive equipment not fully demonstrated under field conditions. The development and use of detection systems for estimating air-borne spore numbers would be a further development in risk assessment for onion downy mildew. This study reports on the development of an immunomonitoring system for conidia of P. destructor.
Materials and methods
Production of P. destructor immunogen for antibody production
The isolate (PD HL00) of P. destructor used in the study was as reported by Gilles et al. (2004). Leaf surface wax material of ten onion sets (Allium cepa cv. White Lisbon) was removed by gentle agitation with sheep’s wool (found to remove leaf wax without leaf damage) prior to inoculation with P. destructor. Twenty-five 20-μl droplets of P. destructor (1 × 104 conidia ml−1 H2O) were applied to each sheep’s wool-treated leaf. To induce infection, inoculated plants were incubated in high humidity for three days after which plants were removed and placed in a temperature-controlled glasshouse (18°C) for a further 2 weeks. Inoculated plants were returned to a high humidity environment for a period of 48 h to induce sporulation by P. destructor on infected leaves.
Collection of P. destructor spores from leaf surfaces
A hand-held Burkard surface cyclone sampler (Burkard Manufacturing Co., Rickmansworth, Herts, UK) was used to collect sporangia of P. destructor from the surface of the infected leaf material. The 2 ml Eppendorf containing the collected spores was removed and 1 ml of chilled sterile distilled water (SDW) was added. The collected P. destructor sporangia were suspended in water and 0.5 ml volume of chilled SDW was added. The sporangial suspension was filtered through a stainless steel membrane (47 μm pore size) to remove any large contaminating material. The liquid phase was collected and bacterial and other small leaf contaminants removed by filtering using a polyester membrane (10 μm pore size). The filtrate was collected and resuspended in 1 ml phosphate buffered saline solution, pH 7.0 (PBS). Bright field microscopy was used to determine the presence of P. destructor sporangia which were adjusted to a concentration of 3.5 × 104 conidia ml−1.
Immunization of mice with P. destructor sporangia
The spore suspension was agitated, using a Gallenkamp spinmix, continuously for a period of 5 min after 3 h at 0–4°C. A microfuge (MSE Microcentaur) was used at 13 rpm for 5 min to separate particulate spore material from the soluble spore fraction of the sample. The soluble fraction of the sample was retained and concentrated at first by freeze-drying (Modulyo 4 k, Edwards) and then rehydrating to a final volume of 100 μl PBS. Two Balb C female mice (coded 7996, 7997) were immunised (by intraperitoneal injection) each with 50 μl of the concentrated soluble P. destructor sporangial preparation mixed with an equal volume of Titermax adjuvant. All further immunisations were as described above. Tail bleeds were taken seven days after the second immunisation procedure and a PTA-ELISA (described below) was carried out to determine whether the mice had produced an immune response to P. destructor.
The mice received a final pre-fusion boost of the P. destructor soluble sporangial immunogen mixed with adjuvant (100 μl). The spleen of mouse 7996 was removed 4 days later and the fusion was carried out according to those methods reported by Dewey (1992). Hybrids were fed on days 3, 6 and 10 and cell culture supernatants screened by PTA ELISA and immunofluorescence 14 days after cell fusion for the presence of antibodies which recognised sporangial components of P. destructor.
Monoclonal antibody screening
Plate trapped antigen ELISA (PTA ELISA)
One hundred μl of P. destructor soluble sporangial washings in PBS were aliquoted in to each of 96-well Polysorp microtitre well strips (Nunc, Roskilde, Denmark; Cat. No.469957). The strips were incubated overnight in an enclosed chamber at 18°C. Unbound material was then removed and the microtitre wells were washed once with 200 μl PBS. The microtitre wells were blocked with 200 μl of 1% Casein buffer (1% (w/v) casein PBS) and incubated at 37°C for 45 min. Residual blocking buffer was removed and wells were washed four times for 1 min each with 200 μl PBS, 0.05 % Tween 20 and 0.1 % Casein (PBSTw C). Each well received 100 μl of fusion hybridoma tissue culture supernatant mixed with PBSTw C. Following incubation in a Wellwarm shaker incubator (30°C) for a period of 45 min as above, wells were washed three times for 1 min each with 200 μl PBSTw. A DAKO duet amplification system was used according to manufacturer’s instructions (DAKO Ltd, Cambridge, UK) to amplify the signal generated by bound tissue culture supernatant antibodies. Wells were washed as described above and 100 μl of 3,3′,5,5′-tetramethylbenzidene substrate (Sigma, UK) was added to each well. The reaction was stopped by adding 25 μl of a 20% 1 M H2S04 solution to each well. Absorbance at 450 nm was determined with a Biohit BP800 ELISA plate reader (Alpha Laboratories, Hampshire, UK).
Immunofluorescence
Twenty microliters of a 1 × 103 spores ml−1 P. destructor conidial spore suspension was aliquoted to individual multiwell glass slides (Cel-Line/Series Scientific Corp, USA; Cat No. 10-3404). Following air drying any unbound spore material was removed with a PBSTwC wash. Material remaining bound to the multiwell glass slides was incubated with 20 μl of hybridoma tissue culture supernatant antibodies (TCS) mixed with PBSTwC for a period of 30 min at room temperature. A counterstain of Evans blue and Eriochrome black was incorporated within the TCS antibody suspension to quench P. destructor spore autoflourescence (Kennedy et al. 1999). Each multiwell received a wash as described above and following air drying was incubated with anti-mouse antibodies which had been conjugated to fluorescein isothiocyanate dye. A counter-stain was included to ensure quenching of sporangial autoflourescence. Incubation was carried out at room temperature in darkness to prevent photo-bleaching of the conjugated antibody. The processed multiwells received a final wash of PBSTwC and after air drying were mounted and viewed by episcopic fluorescence microscopy for the presence of antibody/fluorescein-tagged sporangia of P. destructor. Hybridoma antibody tissue culture supernatants, identified as positive to P. destructor sporangial material using either PTA ELISA and/or IF, were selected and twice cloned to monoclonal Ab status.
Selection of specific P. destructor monoclonals
To determine specificity, the selected P. destructor monoclonal cell lines were determined by PTA-ELISA and IF against a range of fungal species. Tests were carried out on spores and mycelium taken from pure cultures of Bremia lactucae, Peronospora parasitica, Paecilomyces variotii, Botrytis cinerea, B. squamosa, Stemphyllium sp., Aureobasidium pullulans, Phoma betae, Ascochyta rabei, Fusarium culmorum, Penicillium roquefortii, Pyrenophora teres and sporangia of P. destructor. Stains used in these tests are as designated in Kennedy et al. (2000). With the exception of P. destructor, P. parastica, B. lactucae and Ascochyta (all of which were grown directly on plant material) the fungal species used in the reactivity tests were grown on a synthetic medium covered with a sterile Supor membrane filter prior to inoculation. Fourteen days after inoculation (of cultures grown on agar) 5 ml of PBS (pH 7.5) solution was applied to the culture surface. Surface washings were taken by gently stroking the culture surface with a glass spreader. All collected spore concentrations were adjusted to a final concentration of 1 × 105 spores ml−1 PBS. The spore solutions were individually aliquoted into each microtitre well (100 μl per well) of a Polysorp microtitre strip. The wells were covered and incubated overnight at 4°C. Unbound material was removed and the microtitre wells were washed once with 200 μl PBS. An ELISA was carried out as previously described.
Development of a competitive lateral flow assay format for the detection of conidia of P. destructor
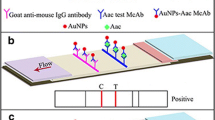
A competitive lateral flow format (Fig. 1), comprising a Millipore 135 HiFlow™ cellulose ester membrane direct cast onto 2 ml Mylar backing (Millipore Corp, USA.), an absorbent pad (Schleicer and Schuell, Germany) and a sample pad (Millipore Corp., USA) was constructed for the detection of P. destructor sporangia. Control lines of an anti-mouse serum were sprayed directly onto the membrane surface using a flat bed air jet dispenser (Biodot Ltd, West Sussex, UK). A collected soluble fraction of a P. destructor sporangial sample, prepared as described earlier, was adjusted to a protein concentration of 500 μg ml−1 , 250 μg ml−1 and 125 μg ml−1 in PBS and applied as a test line again using a flat bed air jet dispenser. Membranes were air-dried at 35°C for a period of 4 h. The test and control line-labelled lateral flows were cut in to 5 mm strips and each strip housed within a plastic case (Schleicer & Schuell, Germany).

Lateral flow cross-section (5 mm strip)
Antibody conjugation
A British Biocell gold anti-mouse IgM solution was pre-mixed (conjugated) with a selected hybridoma cell line (coded EMA 242) before drop application to a 5 mm sample pad and air-drying. Variable concentrations of gold conjugated antibody EMA 242 were applied to different sample pads to investigate the antibody conjugate concentration which gave optional test line formation on the lateral flow device (lfd). Sample conjugate Ab pads were attached to each lfd strip as shown in Fig. 1. A test antigen of a 60 μl sporangial suspension (3 × 103 P. destructor sporangia) was then applied to the sample pad of an lfd strip. The competitive lateral flow devices (clfd) were viewed 5 min post-sample application. For each test, a spore-free suspension was applied to a clfd as a negative control. The variable antibody dilutions of MAb used in these tests are shown in Table 1.
Visual detection threshold of a competitive lateral flow device employing two membrane types for P. destructor sporangia
Studies were carried out using a clfd format for the detection of known concentrations of P. destructor spores. Two different membrane types were examined: a Millipore 135 HiFlow™ cellulose ester membrane direct cast on to 2 ml Mylar backing and a Millipore 240 HiFlow™ cellulose ester cast membrane. The lfd devices were prepared as described above and a test line of 250 μg ml−1 P. destructor soluble antigen in PBS was applied. The membranes were air-dried at 35°C, cut into 5 mm strips and each strip housed within a plastic case as previously described.
A known sporangial concentration of P. destructor sporangia (60 μl) was mixed with EMA 242 gold conjugate (5 μl) to produce a final antibody dilution of either 1:150, 1:400 or 1:600. The mixture was applied to the sample pad of each clfd and results viewed 5 min post-sample application. For each membrane type a ‘spore free suspension’ was mixed with MAb EMA 242 gold conjugate to act as a negative control.
Semi-quantitative tests with lateral flow format using Millipore 135 HiFlow™ cellulose ester membrane
A 5 μl British Biocell gold anti-mouse IgM solution was pre-mixed with 10 μl EMA 242 and then applied drop-wise to lateral flow sample pads at a test volume of 15 μl, each dried as previously described. This was chosen as the optimal concentration for detecting approximately 500 sporangia of P. destructor. Sporangia of P. destructor in sample buffer were applied drop-wise (70 μl) to the sample pads of the pre-prepared lateral flows. Sporangial concentrations ranged from 240 to 960 sporangia applied. The lateral flow devices were viewed 20 min post-sample application for the formation of a test and control line and test line optical density values were generated using a BioDot lateral flow reader (BioDot, Chichester). A negative control of lateral flow running buffer alone (0 downy mildew conidia) was also included within these tests.
Results
Monoclonal antibody screening
Plate trapped antigen ELISA (PTA ELISA)
Eleven hybridoma cell lines were identified (using PTA ELISA) as producing antibodies which recognised components associated with the sporangial material of P. destructor. A preliminary screen against a range of plant fungal pathogens identified three tissue culture supernatants for expansion to monoclonal status. These were selected, cloned to monoclonal antibody status and coded EMA 240, 242, and 243. Monoclonal antibody cell line EMA 240 was not used in reactivity tests as it was observed to react with other downy mildew species when tested by ELISA (data not shown). Monoclonal antibodies EMA 242 and 243 reacted to their homologous antigen (P. destructor), and demonstrated a high level of specificity when tested against other fungal species (Fig. 2).

Reactivity of monoclonal antibodies EMA 242 and 243 to a range of airborne fungal species as tested by PTA ELISA (each value represents the mean of two replications, SD 0.0892)
Immunofluorescence
Only eight of the eleven cell lines were identified as producing antibodies which recognised components directly associated with the conidia of P. destructor when visualised by immunofluorescence. Of these, six were excluded following preliminary reactivity studies (data not shown). Those selected for further testing by IF were EMA 242 and 243 (Table 2). EMA 240 did not react with material directly associated with P. destructor; however, an area of diffuse speckling was noted surrounding the spore. In immunofluorescence studies both EMA 242 and 243 reacted with the spore wall of P. destructor and retained a high level of specificity when tested against other fungal species. This indicated a high degree of similarity between these cell lines.
Assessment of competitive lateral flow assay format for the detection of P. destructor
At a test line application of 500 μg ml−l spore protein deposition, test line inhibition (i.e. no test line development) was observed when a P. destructor spore sample was mixed with gold conjugated EMA 242 at a dilution >1:160. For all negative control samples (i.e. no P. destructor spores present), control and test line development was observed for each competitive lateral flow device. Using an Ab dilution of 1:640 gave no test or control lines for either spore-positive or spore-negative samples.
At a test line concentration of 250 μg ml−l protein (spore) deposition, strong test and control line development was observed at detection antibody (Ab) dilutions of 1:160 and 1:320 when a spore-free suspension was applied. Testing a positive sample of P. destructor and the detection antibody at a dilution of 1:160 gave rise to a barely visible test line, but a strong control line. At an antibody dilution of 1:320, test line depletion was complete. At a test line concentration of 125 μg ml−l , testing a P. destructor spore-free suspension, gave test and control line development when a detection antibody dilution of 1:160 was used. Using a positive P. destructor spore sample gave rise to a clear control line but no test line development (a positive test for the competitive lateral flow format). At all other antibody dilutions, control lines were barely visible and no test line development was noted for any of the samples tested.
Visual detection threshold of a competitive lateral flow device employing two membrane types for P. destructor sporangia
Using a Millipore HiFlow™ 135 Membrane and antibody EMA 242 gold conjugate at a dilution of 1:150, test line formation was observed for all spore samples tested. This denoted that the detection sensitivity of the test was poor and unable to detect 2,000 sporangia of P. destructor. However, by diluting the activity of the antibody conjugate to 1:400, test sensitivity was improved (Table 3a, Fig. 3a). At an antibody conjugate dilution of 1:600, the test became void with no test line formation for any of the samples tested.

Development of a competitive lateral flow for onion downy mildew conidia a competitive lateral flow test employing detection antibody concentration of 1:400, b competitive lateral flow test employing detection antibody concentration of 1:600
Using a Millipore HiFlow™ 240 membrane competitive lateral flow device and EMA 242 conjugated to gold spheres at a dilution of 1:150, test line formation was again observed for all spore samples tested. As previously noted, by diluting the activity of the antibody to 1:400, the sensitivity of the test was improved (Table 3b). For this membrane, an antibody dilution of 1:600 was required to achieve a detection assay where sporangia in excess of 250 could be detected (Fig. 3b, Table 3b).
Semiair-quantitative lateral flow prototype tests with onion downy mildew
The results of using increasing amounts of P. destructor sporangia on the lateral flow device are shown in Table 4. When a negative sample (0 sporangia) was applied to a lateral flow device, strong test line development was observed. As spore concentrations increased, the test line colour formation decreased. When a P. destructor sporangial concentration of 960 was applied to a lateral flow device, no test line development was observed. Using a Bio-dot lateral flow reader, an optical density value of 2.2 was observed with control suspensions (0 sporangia). However, when 960 sporangia of P. destructor was added to the device, the optical density of the line decreased to 0.3.
Discussion
In this study detection tests for P. destructor sporangia were developed although these have not yet been used in the field. Detection of the presence of P. destructor sporangia could be important in onion downy mildew control regimes. Control of plant pathogens could be improved if inoculum could be detected quickly in the field directly by the grower. Airborne inoculum plays a vital role in the development of epidemics caused by Botrytis leaf blight on onion crops (Carisse et al. 2003, 2005). In this work, a linear relationship was found between number of lesions on plants and air-borne Botrytis conidial concentrations. Airborne conidial concentrations of 25 to 35 conidia m−3 of air were associated with 2.5 lesions per leaf. When detection of Botrytis inoculum was used as a control criterion under field conditions, it led to a reduction in fungicide usage of 75 and 56% in 2002 and 2003. A similar relationship between spore number and disease intensity has been reported for Cercospora apii on celery (Berger 1969). In both these studies, microscopes were used to determine spore numbers from air samples.
One of the objectives of the work reported in this paper was to construct rapid tests for sporangia of P. destructor. If rapid tests were suitable for use in the field they could be used potentially to detect sporangia of P. destructor using air samplers as a means of forecasting the onset of disease development. To date no strains of onion downy mildew have been reported in the UK. However it is likely that the antibodies used to construct lateral flow tests developed in this study would react equally with all populations of P. destructor found in the field. By using techniques outlined in this paper, early detection of P. destructor in samples could be made possible. The lateral flow device would, however, need to be tested with portable air samplers in the field to determine the optimal trapping format for P. destructor sporangia. Trapping formats for sporangia would also need to be integrated with numbers of sporangia found above infected crops in the absence of onion downy mildew symptoms on plants. However, by integrating the device with the output from a scanning densitometer, the sensitivity of the device can be improved to enable it to detect the presence of sporangia of P. destructor at lower concentrations. Scanning densitometers are becoming more portable and their use might enable low numbers of P. destructor sporangia to be detected using a lateral flow device. Symptoms of onion downy mildew within crops are difficult to detect at low levels, especially in large cropping areas. Additionally, the symptomology of the disease on young plants is poorly understood and observed. By detecting the presence of P. destructor sporangia, it would be possible to determine action thresholds for onion crops at different stages in their development. The lateral flow device, if used to detect sporangia of P. destructor in the field, would require validation in different onion-producing areas.
Using advanced monitoring techniques, the optimal criteria for applying fungicide applications to an onion crop could be investigated. Disease development might also be detected in the absence of visible symptoms. This is a critical point in considerations of disease control, since if early applications of fungicide can be targeted to when P. destructor sporangia are present, improved control could be achieved. Rapid diagnostic tests, similar to those reported in this study, exist for identifying Phytophthora spp. (Lane et al. 2007). However the test kits reported reacted to a range of Phytophthora spp. which are commonly found in soils. These lateral flow devices and those reported by Thornton et al. (2004) were used to identify infected plant tissues in soil samples. However, in studies reported in this paper, lateral flow devices reacted selectively to P. destructor sporangia (no infected plant material present). If the device were to be used in conjunction with air samples, it would pose fewer problems in comparison to using lateral flow devices for detecting infected soil or plant tissues.
Detecting P. destructor sporangia would be particularly useful early in the season as a method of preventing disease transfer between over-wintered salad onion crops and bulb onions grown as sets or as seeded crops. The use of weekly estimates of inoculum in air samples has also been reported (Kennedy and Wakeham 2006) for other diseases, notably Pyrenopeziza brassicae (light leaf spot of horticultural and arable brassicas). Tests which can be conducted in the field are necessary if information on air-borne inoculum concentration is to be of more practical value. Results of the trials reported in this paper demonstrate the development of lateral flow devices that can detect plant pathogenic inoculum. Potential exists for linking these estimates of P. destructor inoculum to mathematical models describing the environmental factors which affect onion downy mildew sporulation (Gilles et al. 2004). Use of this approach might improve the efficiency of both the inoculum detection system and disease forecasts. However this would require investigation in future work.
References
Battilani, P., Rossi, V., Racca, P., & Giosuè, S. (1996). ONIMIL, a forecaster for primary infection of downy mildew of onion. European Mediterranean Plant Protection Organisation Bulletin, 26, 567–576.
Berger, R. D. (1970). Forecasting Helminthosporium turcicum attacks in Florida sweetcorn. Phytopathology, 60, 1285.
Bugiani, R., Govoni, P., & Cobelli, L. (1998). First large scale application of IPI model for potato late blight prediction in the Po valley. In H. Schepers, & E. Bouma (Eds.), Proceedings of the Workshop on the European Network for the Development of an Integrated Control Strategy of Potato Late Blight (pp. 188–199). Carlow, Ireland: PAV.
Carisse, O., McCartney, H. A., Gagnon, J. A., & Brodeur, L. (2005). Quantification of air-borne inoculum as an aid in the management of leaf blight of onion caused by Botrytis squamosa. Plant Disease, 89, 726–733 doi:10.1094/PD-89-0726.
Carisse, O., Rolland, D., Lefebvre, A., & Talbot, B. (2003). Using aerobiology data to manage onion blight caused by Botrytis squamosa. Proceedings of the 8th International Congress of Plant Pathology, 1, 5.
Clarkson, J. P., Kennedy, R., & Phelps, K. (2000). The effect of temperature and water potential on the production of conidia of sclerotia of Botrytis squamosa. Plant Pathology, 49, 119–128 doi:10.1046/j.1365-3059.2000.00417.x.
Cook, H. T. (1932). Studies on the downy mildew of onion and the causal organism, Peronospora destructor (Berk.) Caspary. New York agricultural experimental. Station, Ithaca, 143, 1–40.
Cruickshank, I. A. M. (1958). Environment and sporulation of phytopathogenic fungi. IV. The effect of light on the formation of conidia of Peronospora tabacina Adam. Australian Journal of Biological Sciences, 16, 87–98.
Day, J. P., Kell, G., & Griffiths, G. W. (2002). Differentiation of Phytophthora infestans from other air-borne biological particles by flow cytometry. Applied and Environmental Microbiology, 68, 37–45 doi:10.1128/AEM.68.1.37-45.2002.
Dewey, F. M. (1992). Detection of plant invading fungi by monoclonal antibodies. In J. M. Duncan, & L. Torrance (Eds.), Techniques for the rapid detection of plant pathogens (pp. 47–62). Oxford: Blackwell.
Gilles, T., Clarkson, J. P., Phelps, K., & Kennedy, R. (2004). Development of MILIONCAST, an improved model for predicting downy mildew sporulation on onions. Plant Disease, 88, 695–702 doi:10.1094/PDIS.2004.88.7.695.
Hildebrand, P. D., & Sutton, J. C. (1982). Weather variables in relation to an epidemic of onion downy mildew. Phytopathology, 72, 219–224.
Jesperson, G. D., & Sutton, J. C. (1987). Evaluation of a forecaster for downy mildew of onion (Allium cepa L.). Crop Protection (Guildford, Surrey), 6, 95–103 doi:10.1016/0261-2194(87)90106-2.
Kennedy, R. (1998). Bulb onions: Evaluation of fungicides for control of downy mildew (Peronospora destructor). Horticultural Development Council, Annual Report (Year 1) for project FV 189, pp. 10.
Kennedy, R., & Wakeham, A. J. (2006). Impact of fungicide resistance on light leaf spot control in vegetable brassicas in Scotland. Aspects of Applied Biology, 78, 51–58.
Kennedy, R., Wakeham, A. J., Byrne, K. G., Meyer, U. M., & Dewey, F. M. (2000). A new method to monitor airborne inoculum of the fungal plant pathogens Mycosphaerella brassicicola and Botrytis cinerea. Applied and Environmental Microbiology, 66, 297–307 doi:10.1128/AEM.66.7.2996-3003.2000.
Kennedy, R., Wakeham, A. J., & Cullington, J. E. (1999). Production and immunodetection of ascospores of Mycosphaerella brassicicola: The ringspot pathogen of vegetable crucifers. Plant Pathology, 48, 297–307 doi:10.1046/j.1365-3059.1999.00341.x.
Lane, C. R., Hobden, E., Walker, L., Barton, V. C., Inman, A. J., Hughes, K. J. D., et al. (2007). Evaluation of a rapid diagnostic field test kit for identification of Phytophthora spp. including P. ramorum and P. kernoviae at the point of inspection. Plant Pathology, 56, 828–835 doi:10.1111/j.1365-3059.2007.01615.x.
Phillon, V. (2003). Timing of sprays against potato late blight, based on daytime average airborne spore concentration. Proceedings of the 8th International Congress of Plant Pathology, 1, 6.
Rondomanski, W. (1967). Studies on the epidemiology of onion downy mildew, Peronospora destructor (Berk.) Fries. Technical Report for 1962–67. Research Institute for Vegetable Crops, Skierniewice, Poland, 23.
Thornton, C. R., Groenhof, A. C., Forrest, R., & Lamotte, R. (2004). A one step immunochromatographic lateral flow device specific to Rhizoctonia solani and certain related species and its use to detect and quantify R. solani in soil. Phytopathology, 94, 280–288 doi:10.1094/PHYTO.2004.94.3.280.
Wakeham, A. J., Kennedy, R., & McCartney, H. A. (2004). Using ELISA to monitor the collection and retention of a range of common airborne spore types in air-samples. Journal of Aerosol Science, 35, 835–850 doi:10.1016/j.jaerosci.2004.01.005.
Williams, R. H., Ward, E., & McCartney, H. A. (2001). Methods of integrating air-sampling and DNA analysis for the detection of airborne fungal spores. Applied and Environmental Microbiology, 67, 2453–2459 doi:10.1128/AEM.67.6.2453-2459.2001.
Yarwood, C. E. (1937). Relation of light to the diurnal periodicity of sporulation of certain downy mildews. Journal of Agricultural Research, 54, 365–373, 8.
Yarwood, C. E. (1943). Onion downy mildew. Hilgardia, 14, 595–691.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kennedy, R., Wakeham, A.J. Development of detection systems for the sporangia of Peronospora destructor . Eur J Plant Pathol 122, 147–155 (2008). https://doi.org/10.1007/s10658-008-9346-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10658-008-9346-6