
Abstract
The concentrations of 16 priority polycyclic aromatic hydrocarbons (PAHs) in four size fractions (<2, 2–20, 20–200, >200 μm) in soils at different depth from a heavily contaminated crude benzol production facility of a coking plant were determined using GC–MS. Vertically, elevated total PAHs concentrations were observed in the soils at 3.0–4.5 m (layer B) and 6.0–7.5 m (layer C), relatively lower at 1.5–3.0 m (layer A) and 10.5–12.0 m (layer D). At all sampling sites, the silt (2–20 μm) contained the highest PAHs concentration (ranged from 726 to 2,711 mg/kg). Despite the substantial change in PAHs concentrations in soils with different particle sizes and lithologies, PAHs composition was similarly dominated by 2–3 ring species (86.5–98.3 %), including acenaphthene, fluorene, and phenanthrene. For the contribution of PAHs mass in each fraction to the bulk soil, the 20–200 μm size fraction had the greatest accumulation of PAHs in loamy sand layers at 1.0–7.5 m, increasing with depth; while in deeper sand layer at 10.5–12.0 m, the >200 μm size fraction showed highest percentages and contributed 81 % of total PAHs mass. For individual PAH distribution, the 2–3 ring PAHs were highly concentrated in the small size fraction (<2 and 2–20 μm); the 4–6 ring PAHs showed the highest concentrations in the 2–20 μm size fraction, increasing with depth. The distribution of PAHs was primarily determined by the sorption on soil organic matter and the characteristics of PAHs. This research should have significant contribution to PAH migration study and remediation design for PAHs-contaminated sites.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Polycyclic aromatic hydrocarbons (PAHs) are compounds consisting of at least two fused benzene rings. These compounds are potentially carcinogenic, mutagenic, and teratogenic and widely exist in the environment as persistent organic pollutants (POPs) (Juhasz and Naidu 2000; Wilcke 2000; Dugay et al. 2002; Meudec et al. 2006). In the late 1970s, the United States Environmental Protection Agency (US EPA) had listed 16 PAHs as priority pollutants, including fluorene (Fle), phenanthrene (Phe), and benzo[a]pyrene (BaP) (Keith and Telliard 1979). PAHs are produced by incomplete combustion or pyrolysis of organic materials. Industrial activities, such as tar and coal gas processing and incomplete combustion of fossil fuels, are the main sources of PAHs distributed into the environment (Mastral and Callén 2000).
Consequently, soils in the industrial sites (e.g., coal fields, coking plants, and natural gas plants) had become a great pool for reserving PAHs and other pollutants (Wild and Jones 1995; Khodadoust et al. 2000; Viglianti et al. 2006). PAHs have low water solubilities, excellent thermal stabilities, and high octanol–water distribution coefficients. Therefore, PAHs can be effectively sorbed onto soil particles and often preferentially concentrated in specific size fractions. Some studies have reported that PAHs are concentrated in larger soil particles (e.g., >125 μm) (Wang et al. 2001; Ahrens and Depree 2004; Oen et al. 2006; Yang et al. 2008). However, Maruya et al. (1996, 2000) have found PAHs concentrated in smaller soil particles such as clay and silt. The different distribution pattern of PAHs in all particle-size fractions is primarily determined by PAHs concentration, soil organic matter (SOM), mineral and clay composition (Hwang and Cutright 2003). The strong adsorption of PAHs onto soil particles could reduce their mobility and bioavailability, thus slowing down their biodegradation, migration, and transformation. It is essential to know the distribution of PAHs in the particle-size fractions in order to understand their physicochemical reactive properties and to effectively design the remediation of PAHs in soil.
A few studies have been conducted to determine the characteristics of PAHs distribution pattern in contaminated soils, mostly at the surface soil (<1 m). These studies also revealed that PAHs are typically concentrated in the surface soils and concentrations of PAHs decreased with depth because of their poor mobility (Krauss et al. 2000; Wang et al. 2001; Li et al. 2010). In this study, we investigated the PAHs distribution in soils from a coking plant in Beijing, China. Our previous work found that, after a coking operation for over 50 years, soils were contaminated by PAHs with a mean local concentration greater than 1,000 mg/kg and PAHs contamination depth reached more than 10 m. As a result, the groundwater at the site was heavily contaminated by PAHs and other pollutants. PAHs distribution may show various patterns in the different depth. Our objectives in this study were to: (1) determine and compare the composition and partitioning of PAHs in different soil particle-size fractions, (2) describe the accumulation, distribution patterns, and migration of the PAHs in the soils located at different depth with different lithology, and (3) analyze the origins of PAHs in soils at different depth. The characteristic of PAHs distribution in the different size fractions would reveal the PAHs sorption behaviors by the soil and thus can provide critical information for the design of remediation plans.
Materials and methods
Soil sample collection
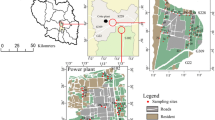
Figure 1 shows the stratum structure of the research area. The area was occupied by a crude benzol production facility. Due to the incomplete combustion of coal and leakage of coke tanks during the production process, soils were contaminated by high concentrations of PAHs (the maximum PAHs concentration at the site was 16,441 mg/kg). Samples were collected from different layers with an impact drill (SH-30 drilling rig, diameter: 130 mm, made in Hebei province, China). Ten sampling wells were selected randomly in a horizontal direction, and four sampling sites were set in a vertical direction of each well (from 0 to 12 m below the ground surface) in the heavily contaminated area to get representative soil samples. There existed four kinds of soil texture at different depth. Samples collected from the same depth and with the same lithology were homogeneously mixed, sealed in pre-baked dark glass jars, and kept at −80 °C before analysis.

Schematic diagram showing the stratum structure of the research area. Lithological characteristics of the sampling soil layers. A 1.5–3.0 m (depth), 27.98–29.48 m (elevation), clay silt; gray, medium density, moist, moldable, containing ash and brick particles, 1.90–2.10 m: cement pavement (hardened). B 3.0–4.5 m (depth), 26.48–27.98 m (elevation), sandy silt; dark gray, medium density, moist, moldable, containing organic matter, texture inhomogeneous. C 6.0–7.5 m (depth), 23.48–24.98 m (elevation), silty sand; brown, medium density, moist, containing mica and iron oxides. D 10.5–12.0 m (depth), 18.98–20.48 m (elevation), fine sand; brown, dense, moist, hard; containing mica and iron oxides
Size separation
The samples were separated into four particle-size fractions following standard methodology (International Soil Science Society 1996) using sonication-sedimentation: clay (<2 μm), silt (2–20 μm), fine sand (20–200 μm), and coarse sand (>200 μm). Briefly, 500 g of sample (on air-dry weight basis) was dispersed by sonication (ultrasonic power: 300 watts, water:soil ratio = 10:1) for 30 min. It was then wet-sieved to pass through a screen (2 mm mesh). The sieved soil was transferred into a 10-L plastic container and then suspended in water at ≤4 % wt and allowed to settle. Each fraction was collected by siphoning at an appropriate sedimentation time determined by Stokes’ law. All soil size fractions were then freeze-dried for chemical analysis and PAHs extraction. Each soil fraction was also weighed. In the size fractionation, 98.9–100 % of the mass of the original soil sample was recovered.
PAHs extraction
Soil samples and corresponding fractions were freeze-dried and sieved to pass a 100-mesh screen. The sieved soil was extracted with dichloromethane under sonication. Two mL of extract was collected and purified using a silica gel column (packing medium: 200–300 mesh silica gel and anhydrous sodium sulfate). The PAHs mixture was eluted with 50 mL of n-hexane:dichloromethane mixture (1:1). The final extract volume was reduced to 1 mL using rotary evaporation; 125 μL of internal standards (2-fluorobiphenyl and p-terphenyl, 2 mg/kg) was added to all extracts prior to chromatographic separation.
GC–MS analysis
The 16 US EPA priority PAHs compounds were detected in the samples using an Agilent 7890A gas chromatography (GC) coupled with a 5975C mass spectroscopy (MS) system in selected ion monitoring (SIM) mode. A capillary column (30 m × 0.25 mm i.d., 0.25 μm film thickness) was used for the determination of PAHs. The column temperature was initiated at 50 °C and increased to 200 °C at 10 °C/min, 280 °C at 5 °C/min (held for 10 min), and 300 °C at 10 °C/min. Injection of 1-μL samples was conducted in the splitless mode. High-purity helium (99.999 %) was use as carrier gas at a linear velocity of 37 cm/s. The pre-column pressure was 0.03 MPa, and front inlet and MS detector temperature were both fixed at 300 °C. The PAHs were separated by GC and then analyzed using a coupled quadrupole mass spectrometer operating under the following conditions: ion source and quadrupole temperatures were respectively set to 230 and 150 °C, multiplier voltage: 1,600 V, interface temperature: 200 °C, ionization energy: 70 eV, scan range: 50–500 amu, and a scan rate: 1.43 scans/s.
TOC and TN analysis
The total organic carbon (TOC) in the soil samples and each size fraction was measured using a TOC analyzer (Liquid II series), and the amount of total nitrogen (TN) was measured using the Kjeldahl method.
Quality control
All experiments were carried out in triplicates. The variation of PAHs concentrations of triplicate soil samples was less than 10 %. Deuterated PAH surrogate standards (phenanthrene-d10, chrysene-d12, and perylene-d12) were added into soil samples to monitor the procedures of sample extraction, clean-up and analysis. Recoveries of three deuterated surrogates added to the soil samples were 82 % for phenanthrene-d10, 81 % for chrysene-d12, and 119 % (n = 7) for perylene-d12, respectively. All the values reported in this paper were not corrected to achieve 100 % recovery.
Results and discussion
The PAHs distribution in soil at different depth
Concentrations of PAHs in soils at different depth varied substantially (Table 1). Soil composition, PAH properties, and leaching are important influential factors for the vertical migration of PAHs in soils (Krauss et al. 2000). A few studies revealed that due to the low mobility of PAHs, their concentrations decreased with depth (Wang et al. 2001; Li et al. 2010). In our study, the total PAHs (TPAH) concentration was the highest at 3.0–4.5 m (layer B) and 6.0–7.5 m (layer C) and relatively lower at both 1.5–3.0 m (layer A) and 10.5–12.0 m (layer D). The 2–3 ring PAHs such as Ane, Fle, and Phe accounted for 86.5–98.3 % of TPAH concentration. The distribution pattern of Ane, Fle in the samples was consistent with TPAH concentration pattern. The ratio of Phe/Ant was about 10:1 at each depth, and either the Phe or Ant concentrations in layer B, C, D were similar to each other: 3–4 times of the concentrations in layer A. The concentrations of the 4–6 ring PAHs were very low, increasing with depth; the concentrations of high molecular PAHs (e.g., Bbf, BaP, IcdP, DahA, BghiP) in the deeper layer (D) were 60–90 times higher than their respective concentrations in layer A.
This unusual distribution pattern may be attributed to the following factors: (1) PAHs with small molecular weights migrate easily. Therefore, small molecule PAHs at the layer A or layer D may be lost to the atmosphere by evaporation or to the groundwater due to pollutants migration and exchanges. (2) There are a silt clay layer and a clayey silt layer located at depth of 7.70–8.30 and 8.30–9.50 m, respectively. They are both characterized by fine soil particles and high densities. These layers may prevent the PAHs from migrating down to the deeper layer D. (3) The TOC content may affect the PAHs sorption to a certain extent. The TOC content in layer A, B, C, D was 11.65, 12.71, 9.50, 5.49 %, respectively. PAHs are preferentially adsorbed onto components rich in organic matter in soil (Huang et al. 1996; Maruya et al. 1996). For example, gravels had lower PAHs sorption capacities and strengths than soils because of their lower organic matter concentrations (Weissenfels et al. 1992). The TOC content was consistent with TPAH concentrations at different depth except the layer A. Although the layer A held a high TOC content, it may enhance the microbial degradation of the PAHs due to well aeration and sufficient oxygen, and PAHs may release due to evaporation. (4) PAHs showed a high concentration in layer D. Base on our preliminary results, the natural dynamic type of the interlayer diving was infiltrate-runoff type, and the groundwater level was mainly from meteoric recharge and groundwater lateral recharge. The dynamic changes of groundwater level generally showed higher from November to March, and relatively low in other months, with a natural changes in the annual rate of 1 m (due to less impact of artisanal mining, the groundwater level changed less). Consequently, the high tar oil contamination in groundwater may affect the high molecular PAHs content in the layer D.
Total PAHs concentrations and PAHs mass in different particle-size fractions
According to international system of soil texture classification standards, layers A, B, and C were all loamy sand, and layer D was sand. The soil in layers A, B, and C contained mostly (73–85 %) fine sand (20–200 μm) (Table 2). The layer D contained >85 % coarse sand (>200 μm). In comparison with the other fractions, the clay (<2 μm) made up only a small proportion (0.62–2.92 %) of the soil mass.
The silt (2–20 μm) had the highest TPAH concentrations, and clay (<2 μm) had higher TPAH concentration than both the fine and coarse sand fractions in most soil layers in this coking plant (except layer A). The PAHs distribution patterns of each particle-size fraction in the same soil texture were different. In layer A, the TPAH concentrations were silt > coarse sand > fine sand > clay; In B, C, and D layers, the TPAH concentrations followed the same trend, being: silt > clay > fine sand > coarse and TPAH concentration decreased with increasing particle size (except for the <2 μm fraction); In layer D, the 2–20 μm size fraction also had the highest TPAH concentrations close to layer A.
PAHs were mainly concentrated in small particle-size fractions (<2 and 2–20 μm). This effect has been widely studied. Maruya et al. (1996) also found that the smaller particles (such as clay) had higher PAHs sorption than the fine and coarse sands. Müller et al. (2000) studied some urban topsoils (0–50 mm) in Bangkok (Thailand) and observed that silt (2–20 μm) and clay (<2 μm) had higher affinity for PAHs than the fine (20–250 μm) and coarse sands (250–2,000 μm). Krauss and Wilcke (2002) likewise investigated the characteristics of PAHs distribution in the particle-size fractions of 11 urban topsoils and found that the PAHs concentration followed the order of: silt (2–20 μm) > clay (<2 μm) > fine sand (20–250 μm) > coarse sand (250–2,000 μm).
PAHs are a family of hydrophobic pollutants, and organic matter in soil is one of the major influential factors of PAHs adsorption onto soil particles. PAHs can be strongly sorbed on soil and difficult to be degraded. They can remain in the environment for a long time. Two factors may be responsible for the higher concentration of PAHs in small soil particles observed in our study. First, because PAHs are non-polar and hydrophobic, the distribution of PAHs in soils is primarily determined by the degree of sorption by the SOM. TOC is likely to act as a carrier for the vertical migration of PAHs in soils, because PAHs migration was mainly associated with organic particles (Bogan and Sullivan 2003). In our study, the smaller-particle-size fractions had substantial higher TOC than the larger ones (fine and coarse sands). With exception of the clay fraction, the patterns of TPAH variation in each soil layer were similar to that of the TOC. The TOC and TOC/TN ratio also suggested that the organic matter in the fine and coarse sands was mainly plant detritus, which should have a high concentration of carbohydrates present and can be easily degraded by microorganisms. Consequently, the large-particle-size fractions sorbed less PAHs. Additionally, some studies revealed that the organic matter present in the silt and clay had a higher degrees of humification and consisted mainly of aromatic/aliphatic hydrocarbons including PAHs that are difficult for soil microorganisms to degrade (Krauss and Wilcke 2002; Gunasekara and Xing 2003). PAHs, as non-polar compounds, have a high affinity for hydrophobic substances. Therefore, as determined by the presence of non-degradable hydrophilic organic matter in soil, PAHs are preferentially distributed in smaller soil particles relative to larger ones. Second, smaller particles are characterized by a larger specific surface area, abundant surface charge, and a higher organic matter density. They have more sorption sites and can provide a larger sorption capacity for PAHs correspondingly (Amellal et al. 2001). Comparatively, larger particles (e.g., fine sand), although accounting for a large fraction of the soil weight, could not effectively capture PAHs because of their smaller specific surface area. Other researchers found a high concentration of PAHs sorbed onto soil particles in the size range of 125–250, 250–500, and >500 μm fractions and suggested that the differences in organic carbon (OC) and black carbon (BC) content partially caused the preferential distribution of PAHs in the different particle-size fractions (Simpson et al. 1998; Wang et al. 2001; Ahrens and Depree 2004; Ni et al. 2006; Oen et al. 2006; Yang et al. 2008; Li et al. 2010).
Overall, the mass distribution of PAHs in the soil stratum is determined by their concentrations in particle-size fractions and the mass contribution of each fraction to the soil. The samples collected from different layers showed a wide variation in fraction composition (Fig. 2) as a result of their different lithology. The 20–200 μm size fraction of layers A, B, and C had low PAHs concentrations, but it contained most of the PAH mass because of the large proportions (73–84 %) of this fraction in the total soil mass. The 20–200 μm size fraction contained 55, 71, and 77 % of PAHs in layers A, B, and C, respectively, revealing an increasing degree of PAHs partitioning with depth. Conversely, the PAHs mass partitioning in the 2–20 μm size fraction decreased with depth, being 32, 22 and 7 % in layers A, B, and C, respectively. In layer D, the >200 μm size fraction contained 81 % of the total PAHs mass followed by the 20–200 μm size fraction (13 %). As a result, the 20–200 μm size fraction contained the most PAHs mass in the 0–10 m soils. Comparatively, in the 10–12 m soils, >200 μm size fraction contained the most PAHs mass. Although the 2–20 μm size fraction had the highest TPAH concentration regardless of sampling depth, its mass contribution was small. Determination of these mass partitioning patterns is highly beneficial to the design of prospective remediation. For example, the concentrations of PAHs in the coarse and fine sand were quite high, while these sand particles have relatively low sorption for PAHs and can easily be removed. Therefore, the predominant association of PAHs with the larger soil particle fractions may be favorable for the future remediation of this coking plant. Richard and Berit (1999) also reported contaminants concentrating into a relatively small mass of fine-grained material, and they suggested that soil particle-size separation should be managed to reduce the volume of contaminated material before soil washing remediation. Therefore, our finding enables selective treatment of fine-grained fractions in the remediation operations for PAHs-contaminated sites.

Percentage of PAHs mass in the different soil fractions
Individual PAH concentrations in different size fractions
We further analyzed the concentrations of the 16 priority PAH pollutants in different particle-size fractions (Fig. 3). We found that the PAHs mainly consisted of 2–3 ring species and a small proportion of 4–6 ring species (10–1,000 times less), consistent with the patterns of TPAH distribution in each soil layer. The 2–3 ring PAHs in the samples consisted of mostly Ane and Fle. Ane and Phe were highly enriched in small size fractions, which was the same pattern observed in the TPAH concentrations. The highest concentrations of the 4–6 ring PAHs occurred in the 2–20 μm fraction of all soil layers, and the concentration of the 4–6 ring PAHs in all particle-size fractions increased with sampling depth.

Sixteen types of PAH concentrations in the different soil fractions
The distribution of PAHs in the particle-size fractions of a soil was determined by the properties of these species and also by the amount of organic matter present. This was demonstrated by the distribution of Phe (a strong acute toxicant) and BaP (a strong genotoxicant). Phe was poorly soluble in water and characterized by a high octanol/water partitioning coefficient and a low vapor pressure. It was also highly mobile in the environment, carcinogenic to animals, and an irritant and an allergen when it contacts human skin. In layer A, the highest Phe concentration occurred in the 2–20 μm fraction, followed by the >200 μm fraction. The lowest concentration was in the <2 μm fraction. In layers B and C, the greatest concentration of Phe was found in the 2–20 μm fraction (4–6 times greater than those in the <2 μm fraction and 20–200 μm fraction), and the lowest concentration was in the >200 μm fraction. In layer D, Phe had similar concentrations in all four particle-size fractions, with the highest concentration in the 2–20 μm fraction. At the coking plant, Phe was preferentially distributed in the silt because of its strong affinity to TOC. Ni et al. (2008) studied the Phe contamination in surface farmland soil collected from Yangtze River delta (China) and found that the highest Phe concentration was in the coarse sand and the lowest concentration in the coarse silt, consistent with the distribution pattern of SOM. Benzo(a)pyrene is also insoluble in water and easily associated with organic colloids. It tends to stay in soil and is relatively immobile in the environment. As a recalcitrant aromatic pollutant, it is often considered as the representative of PAHs emission from coking plants. Moreover, BaP is strong carcinogenic and can induce cancer in the epithelial tissues, such as the skin, lungs, stomach, and digestive tract. Our analysis has revealed that, in layers A and B, BaP had the highest concentrations in the 2–20 μm fractions and the lowest concentrations in the <2 μm fractions. The concentrations in the 20–200 μm fractions and >200 μm fractions were similar. In layers C and D, the BaP concentrations were in this order: silt > fine sand > clay > coarse sand. Furthermore, the BaP concentration in the 2–20 μm fraction increased with sampling depth (layers A–D: 2.06, 6.76, 10.84, and 19.83 mg/kg, respectively). Means (1980) compared the partitioning coefficients of PAHs in 14 soils and sediments, and observed that BaP was associated with the SOM. Sequarisa et al. (2005) analyzed BaP partitioning in an arable topsoil and found that it followed a pattern (in descending order of): >20, 2–20, 0.05–2, <0.05 μm. All these findings showed that BaP tended to be associated with small particles and could be strongly sorbed by SOM.
The distribution patterns of the 2–3 ring and 4–6 ring PAHs in different size soil particles were analyzed. All the 2–3 ring/4–6 ring ratios ranged between 2 and 70, with 2–3 ring PAHs accounting for 68–99 % of the TPAH. This indicated that the 2–3 ring PAHs made up the majority of the TPAH in the soil regardless of sampling depth and soil particle size. In addition, the 4–6 ring PAHs concentration increased with depth. For each particle-size fraction, the 2–3 ring/4–6 ring ratio decreased with sampling depth. The 2–3 ring PAHs were more predominant in the layers A, B and C, and the 4–6 ring PAH concentrations were higher in the layer D than other layers. In all layers, the <2 μm fraction showed a greater concentration of PAHs than the other three fractions, suggesting that the <2 μm fraction had a higher sorption capacity for the 2–3 ring PAHs. The finding is inconsistent with two previous studies (Krauss et al. 2000; Biache et al. 2008). Krauss et al. (2000) observed that the 2–3 ring PAHs were predominantly concentrated in the deeper soil and the 4–6 ring PAHs, being poorly soluble in water, mainly existed in the top and subsurface soils and could not migrate to the deeper soils. Kubiki and Apitz (1999) suggested that the high molecular PAHs might be associated with SOM via strong π–π interactions and thereby were prevented from migration. Thiele and Brüümmer (2002) discovered that the concentration of 4–6 ring PAHs in soil might increase under oxygen deficient conditions due to the biological formation of PAHs from plant material and humid substances, consistent with our findings in this study.
Sources of PAHs in soils of the study area
PAHs from different sources exhibit different distribution patterns in soils, and the knowledge of their origins is critical for the evaluation of their environmental impact. Normally, high molecular weight PAHs are mainly derived from pyrolytic sources, and low molecular weight PAHs are mainly derived from petrogenic sources (Neff et al. 2003). Currently, PAHs are usually tracked for their sources by their species ratios. Ant/(Ant + Phe), Fla/(Fla + Pyr), Phe/Ant and Fla/Pyr ratios can be used to distinguish PAHs source from pyrolysis and petroleum (Budzinski et al. 1997; Yunker et al. 2002).
In this study, the Ant/(Ant + Phe) and Fla/(Fla + Pyr) ratios were analyzed to determine the dominant source of PAHs associated with different soil size fractions at four different depth. For all four layers, the Fla/(Fla + Pyr) ratios in most of the particle-size fractions were >0.5, and the Fla/Pyr ratio in the bulk soil was close to 1.4 (1.49–1.80), confirming the pyrolytic origin of PAHs in the soil. All particle-size fractions of the layer A showed Ant/(Ant + Phe) ratios <0.1 (Fig. 4), and the bulk soil had Phe/Ant ratios >15, suggesting that the PAHs in this layer were originated from leakage or emission during manufacturing or transportation of tar oil, which was one of the main products in a coking plant. With the exception of the clay fraction, other fractions in layers B, C, and D had Ant/(Ant + Phe) ratios >0.1 and the bulk soil had Phe/Ant ratios <10, suggesting that the PAHs in these three layers were originated from incomplete combustion of fuels such as coal. These petroleum-derived low molecular weight PAHs are degraded more rapidly (Jones et al. 1986); relatively, the 3–4 ring PAHs are more difficult to degrade and maintain their source feature (Sauer and Boehm 1991). The high molecular weight PAHs derived from pyrolytic origin are adsorbed on black carbon and difficult to degrade (Muel and Saguem 1985; Nam et al. 2008). In addition, the physicochemical properties of PAHs, such as photo-oxidation, can modify the original distribution characteristics of the sources to some extent (Butler and Crossley 1981). Overall, these data showed that the major source of PAHs contamination in the research area was from incomplete combustion of fuels such as coal (or pyrolytic sources), and from the contamination of the layer A by industrial operations with compounds such as petroleum-related products (or petrogenic sources).

Plots of the ratios of Ant/(Ant + Phe) and Fla/(Fla + Pyr) in the different particle-size fractions of the four soil layers for diagnosing the sources of the PAH present
Conclusions
The results obtained in this work increased our knowledge on the PAHs concentrations, distribution, and possible sources in different particle-size fractions in the contaminated soils at different depth. The PAHs distribution patterns suggested that PAHs in all soils were dominated by the 2–3 ring PAHs (including Ane, Fle, Phe). PAHs concentration was higher in soils at 3.0–4.5 and 6.0–7.5 m, relatively lower at 1.5–3.0 and 10.5–12.0 m. The concentration of TPAH of soil at 1.5–3.0 m declined in order of: silt > coarse sand > fine sand > clay, while soils at 3.0–12.0 m showed the opposite trend. The variations in TOC and TN with particle size followed the trends of the TPAH (except for the clay fraction).
PAHs were presented in higher concentrations in the 2–20 μm size fraction. In the loamy sand soils, the mass of PAHs accumulation was the highest in the 20–200 μm size fraction and increased with depth, while the mass of PAHs in 2–20 μm soil fraction decreased with depth. In the sandy soil, the PAHs mass accumulation was the highest in the >200 μm fraction. For individual PAH distribution, the 2–3 ring PAHs were present at the highest concentrations in the 2–20 μm size fraction in loamy sand layers, and highly concentrated in the <2 μm size fraction in the sand layer. The 4–6 ring PAHs were concentrated in the 2–20 μm size fraction, and their concentrations in all particle-size fractions increased with depth. Based on the Ant/(Ant + Phe) and Fla/(Fla + Pyr) ratio values in soils, it can be concluded that the major source of PAHs in the research area was pyrolytic, and the contamination of soils at 1.5–3.0 m was from petrogenic sources.
References
Ahrens, M. J., & Depree, C. V. (2004). Inhomogeneous distribution of polycyclic aromatic hydrocarbons in different size and density fractions of contaminated sediment from Auckland harbor, New Zealand: An opportunity for mitigation. Marine Pollution Bulletin, 48, 341–350.
Amellal, N., Portal, J. M., & Berthelin, J. (2001). Effect of soil structure on the bioavailability of polycyclic aromatic hydrocarbons within aggregates of a contaminated soil. Applied Geochemistry, 16, 1611–1619.
Biache, C., Laurence, M. H., Faure, P., Colette, M. L., & Leyvalb, C. (2008). Effects of thermal desorption on the composition of two coking plant soils: Impact on solvent extractable organic compounds and metal bioavailability. Environmental Pollution, 156, 671–677.
Bogan, B. W., & Sullivan, W. R. (2003). Physicochemical soil parameters affecting sequestration and mycobacterial biodegradation of polycyclic aromatic hydrocarbons in soil. Chemosphere, 52, 1717–1726.
Budzinski, H., Jones, I., Bellocq, J., Pierard, C., & Garrigues, P. (1997). Evaluation of sediment contamination by polycyclic aromatic hydrocarbons in the Gironde estuary. Marine Chemistry, 58, 85–97.
Butler, J. D., & Crossley, F. (1981). Reactivity of polycyclic aromatic hydrocarbons adsorbed on soot particles. Atmosphere Environment, 15, 91–94.
Dugay, A., Herrenknecht, C., Czok, M., Guyon, F., & Pages, N. (2002). Newprocedure for selective extraction of polycyclic aromatic hydrocarbons in plants for gas chromatographic—mass spectrometric analysis [J]. Journal of Chromatography A, 958, 1–7.
Gunasekara, A. S., & Xing, B. S. (2003). Sorption and desorption of naphthalene by soil organic matter. Journal of Environmental Quality, 32, 240–246.
Huang, W. L., Schlautman, M. A., & Weber, W. J. (1996). A distributed reactivity model for sorption by soils and sediments: The influence of near-surface characteristics in mineral domains [J]. Environmental Science and Technology, 30, 2993–3000.
Hwang, S., & Cutright, T. J. (2003). Statistical implications of pyrene and phenanthrene sorptive phenomena: Effects of sorbent and solute properties. Archives of Environment Contamination Toxicity, 44, 152–159.
Jones, D. M., Rowland, S. J., Douglas, A. G., & Howells, S. (1986). An examination of the fate of Nigerian crude oil in surface sediments of the Humber estuary by gas chromatography and gas chromatography-mass spectrometry. Environmental Analytical Chemistry, 24, 227–247.
Juhasz, A. L., & Naidu, R. (2000). Bioremediation of high molecular weight polycyclic aromatic hydrocarbons: A review of the microbial degradation of benzo[a] pyrene. International Biodeterioration and Biodegradation, 45, 57–88.
Keith, L. H., & Telliard, W. A. (1979). Priority pollutants: I-A perspective view. Environmental Science and Technology, 13(4), 416–423.
Khodadoust, A. P., Bagchi, R., Suidan, M. T., Brenner, R. C., & Sellers, N. G. (2000). Removal of PAHs from highly contaminated soils found at prior manufactured gas operations, Journal of Hazardous Materials(B), 80, 159–174.
Krauss, M., & Wilcke, W. (2002). Sorption strength of persistent organic pollutants in particle size fractions of urban soils[J]. Soil Science Society of America, 66, 430–437.
Krauss, M., Wilcke, W., & Zech, W. (2000). Polycyclic aromatic hydrocarbons and polychlorinated biphenyls in forest soils: Depth distribution as indicator of different fate [J]. Environmental Pollution, 110, 79–88.
Kubiki, J. D., & Apitz, S. E. (1999). Model of natural organic matter and interactions with organic contaminants[J]. Organic Geochemistry, 30, 911–927.
Li, H. L., Chen, J. J., Wu, W., & Piao, X. S. (2010). Distribution of polycyclic aromatic hydrocarbons in different size fractions of soil from a coke oven plant and its relationship to organic carbon content. Journal of Hazardous Materials, 176, 729–734.
Maruya, K. A., Risebrough, R. W., & Horne, A. J. (1996). Partitioning of polynuclear aromatic hydrocarbons between sediments from San Francisco Bay and their porewater. Environmental Science and Technology, 30, 2942–2947.
Mastral, A. M., & Callén, M. S. (2000). A review on polycyclic aromatic hydrocarbon (PAH) emissions from energy generation[J]. Environmental Science and Technology, 34(15), 3051–3057.
Means, J. C. (1980). Sorption of polycyclic aromatic hydrocarbons by sediments and soils [J]. Environment Science and Technology, 14(12), 1524–1528.
Meudec, A., Dussauze, J., Jourdin, M., Deslandes, E., & Poupart, N. (2006). Gas chorolatographic-mass spectrometric method for polycyclic aromatic hydrocarbon analysis in plant biota[J]. Journal of Chromatography A, 1108, 240–247.
Muel, B., & Saguem, S. (1985). Determination of 23 polycyclic aromatic hydrocarbons in atmospheric particulate matter of the Paris area and photolysis by sunlight. International Journal of Environmental Analytical Chemistry, 19, 111–131.
Müller, S., Wilcke, W., Kanchanakool, N., & Zech, W. (2000). Polycyclic aromatic hydrocarbons (PAHs) and polychlorinated biphenyls (PCBs) in particle size separates of urban soils in Bangkok, Thailand. Soil Science, 165, 412–419.
Nam, J. J., Gustafsson, Ö., Kurt-Karakus, P., Breivik, K., Steinnes, E., & Jones, K. C. (2008). Relationships between organic matter, black carbon and persistent organic pollutants in European background soils: Implications for sources and environment fate. Environmental Pollution, 156, 809–817.
Neff, I. M., Boehm, P. D., Kropp, R., Stubblefield, W. A., & Pages, D. S. (2003). Monitoring recovery of Prince William Sound, Alaska, following the Exxon Valdezoil spill: Bioavailability of PAH in offshore sediments. In Proceedings of the international oil spill conference, Publication No. I4730 B. Washington. DC: American Petroleum Institute.
Ni, J. Z., Luo, Y. M., Wei, R., & Li, X. H. (2008). Distribution patterns of polycyclic aromatic hydrocarbons among different organic carbon fractions of polluted agricultural soils. Geoderma, 146, 277–282.
Ni, J. Z., Luo, Y. M., & Zhang, C. B. (2006). Soil environmental quality and remediation in Yangze river delta region: Distribution characteristics of Phenanthrene and Benzo[a]pyrene in particle size separates of agricultural soils[J]. Acta Pedologica Sinica, 43(5), 717–722.
Oen, A. M. P., Gerard, C., & Gijs, D. B. (2006). Relation between PAH and black carbon contents in size fractions of Norwegian harbor sediments. Environmental Pollution, 141, 370–380.
Richard, G. S., & Berit, A. B. (1999). Laboratory treatability testing of soils contaminated with lead and PCBs using particle-size separation and soil washing. Journal of Hazardous Materials, 66, 137–150.
Sauer, T. C., & Boehm, P. D. (1991). The use of defensible analytical chemical measurements for oil spill natural resource damage assessment. In Proceeding of the 1991 international oil spill conference, San Diego, CA, March 4–7, USCG, API, and EPA, pp. 363–369.
Sequarisa, J. M., Lavorentib, A., & Burauela, P. (2005). Equilibrium partitioning of 14C-benzo(a) pyrene and 14 C-benazolin between fractionated phases from an arable topsoil. Environmental Pollution, 135, 491–500.
Simpson, C. D., Harrington, C. F., & Cullen, W. R. (1998). Polycyclic aromatic hydrocarbon contamination in marine sediments near Kitimat, British Columbia. Environmental Science and Technology, 32, 3266–3272.
Thiele, S., & Brüümmer, G. W. (2002). Bioformation of polycyclic aromatic hydrocarbons in soil under oxygen deficient conditions. Soil Biology & Biochemistry, 34, 733–735.
Viglianti, C., Hanna, K., & Brauer, C. (2006). Removal of polycyclic aromatic hydrocarbons from aged-contaminated soil using cyclodextrins: Experimental study. Environmental Pollution, 140, 427–435.
Wang, X. C., Zhang, Y. X., & Chen, R. F. (2001). Distribution and partitioning of polycyclic aromatic hydrocarbons (PAHs) in different size fractions in sediments from Boston harbor, United States. Marine Pollution Bulletin, 42, 1139–1149.
Weissenfels, W. D., Klewer, H. J., & Langhoff, J. (1992). Adsorption of polycyclic aromatic hydrocarbons (PAHs) by soil particles: Influence on biodegradability and biotoxicity. Application Microbiol Biotechnolohy, 36, 689–696.
Wilcke, W. (2000). Polycyclic aromatic hydrocarbons (PAHs) in soil: A review. Journal of Plant Nutrient and Soil Science, 163, 229–248.
Wild, S. R., & Jones, K. C. (1995). Polynuclear aromatic hydrocarbons in the United Kingdom environment: A preliminary source inventory and budget. Environmental Pollution, 88, 91–108.
Yang, Y., Bertrand, L., Carmen, P., Peter, G., & Thilo, H. (2008). Occurrence of coal and coal-derived particle-bound polycyclic aromatic hydrocarbons (PAHs) in a river flood plain soil. Environmental Pollution, 151, 121–129.
Yunker, M. B., Macdonald, R. W., Vingarzan, R., Mitchell, R. H., Goyette, D., & Sylvestre, S. (2002). PAHs in the Fraser river basin: A critical appraisal of PAH ratios as indicators of PAH sources and composition. Organic Geochemistry, 33, 489–515.
Acknowledgments
The authors thank the assistance from Dr. Zhong, L. R. of the Pacific Northwest National Laboratory of USA, and the reviewers of the manuscript for their helpful comments and discussions regarding the work. This work was financially supported by the National High Technology Research and Development Program of China (863 Program) (2009AA063102 and 2012AA06A201), the Science and Technology Key Projects of Beijing, China (D08040900360803).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Liao, X., Ma, D., Yan, X. et al. Distribution pattern of polycyclic aromatic hydrocarbons in particle-size fractions of coking plant soils from different depth. Environ Geochem Health 35, 271–282 (2013). https://doi.org/10.1007/s10653-012-9482-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10653-012-9482-y