
Abstract
The influence of ionic strength, index cations and competing anions on arsenate (AsV) adsorption–desorption kinetics was studied in an Alfisol soil. A flow-through reactor system similar to that developed by Carski and Sparks (Soil Sci Soc Am J 49:1114–1116, 1985) was constructed for the experiments. Arsenate adsorption kinetics for all the treatments were initially fast with 58–91% of AsV adsorbed in the first 15 min. Beyond 15 min, AsV adsorption continued at a slower rate for the observation period of the experiments. Changes in the solution composition had differing effects on the cumulative amount of AsV adsorbed by the soil. Ionic strength and different index cations had little effect on the amount of AsV adsorbed, while the presence of phosphate decreased the amount of AsV adsorbed from 169 to 89 and 177 to 115 g AsV μg−1 in 0.03 M sodium nitrate and 0.01 M calcium nitrate, respectively. Considerably less AsV was desorbed than was adsorbed, with only between 2 to 17% of the adsorbed AsV desorbed. The presence of phosphate increased the amount of AsV desorbed by 17%, but other changes in the solution ionic strength or index cation had little effect on the amount of AsV desorbed.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Arsenic (As) sorption on pure mineral and soil surfaces has been extensively studied to evaluate its mobility and fate in soils, surface and groundwater. Sorption reactions have been the focus of these studies, as in most cases, these processes control the concentration of As in soil solutions and therefore the bioavailability of As in the environment (Fendorf et al. 1997). Although the extent of adsorption and desorption of As by mineral surfaces (Anderson et al. 1976; Pierce and Moore 1980; Manning and Goldberg 1997a; Grafe and Sparks 2005) and soils (Manning and Goldberg 1997b; Smith et al. 1999) has been widely studied, limited information is available on the kinetics of the sorption reactions and the factors that influence such reactions. Knowledge of sorption reaction rates is useful for providing an insight into reaction mechanisms and the processes occurring on the adsorbing surface (Skopp 1986).
In most studies, information on the kinetics of As sorption has been obtained using batch methods (e.g., Anderson et al. 1976; Pierce and Moore 1980; Fuller et al. 1993; Scott and Morgan 1995; Raven et al. 1998). Based on such studies, numerous researchers have reported that the adsorption of both arsenate (AsV) and arsenite (AsIII) on pure minerals and soils is rapid (Anderson et al. 1976; Pierce and Moore 1980; Raven et al. 1998). For example, Elkhatib et al. (1984) reported that the initial reaction of AsIII with five surface and subsurface soils that had a range of chemical and physical properties (coarse-loamy, mixed, mesic Typic Hapludults; fine-loamy, mixed, mesic Typic Hapludults; coarse-loamy, mixed, mesic Fluventic Dystrochrepts; fine, mixed, mesic Typic Hapludalfs) was rapid, with more than 50% (5–500 mg AsIII l−1) of the original AsIII being adsorbed in the first 30 min. Raven et al. (1998) also observed that the adsorption of both AsV and AsIII on ferrihydrite was rapid, with AsIII adsorption being considerably faster (i.e., achieved equilibrium) than AsV at the concentrations studied. The higher rate of AsIII adsorption may be related to the nature of the adsorption mechanism of the ions and the surface coverage of the As species on the ferrihydrite surface (Raven et al. 1998). After 2 h, they found that 99.7 and 98.3% of AsIII and 79.6 and 83.4% of AsV were adsorbed at pH 4.6 and 9.2, respectively, and it was considered that complete adsorption occurred after 96 h. Although batch methods provide valuable insight into chemical reactions at equilibrium (Skopp 1986), there are several disadvantages with these techniques. A major drawback of batch techniques is that they are closed systems, and desorbed species are not removed from the system being studied (Sparks 1989; Harter 1991). The presence of the desorbed species in the batch reactor may inhibit desorption of ions from the solid phase (Sparks 1989; Amacher 1991). Data interpretation is therefore difficult, as reverse reactions must be taken into account, unless there is only one reaction and it is unidirectional (Amacher 1991). An alternative method to the batch technique that significantly decreases reverse reactions by continually removing solution from the reaction chamber and preventing the accumulation of desorbed species is the stirred-flow method. The stirred-flow technique has been used to study time-dependent sorption reactions (Carski and Sparks 1985; Seyfried et al. 1989; Toner et al. 1989; Bar-Tal et al. 1995; Yin et al. 1997). Furthermore, the stirred-flow technique exposes the adsorbing surfaces to a constant solution concentration of adsorbent that is continually being removed until the easily accessible sorption sites are saturated, allowing the apparent reaction rate to be measured.
In previous published papers (Smith et al. 1999, 2002), research was focused on the interactions of As in the soil–solution environment and identified several parameters, such as pH, ionic strength (I), index cation and competing anions, that influence As adsorption. In this study we investigated the effects of I, index cations and other anions on the kinetics of AsV sorption. Such information is lacking in the literature as most kinetic studies to date have focussed on the adsorption from soils in constant background electrolyte solutions. Few studies have investigated the effects of ionic strength (I), index cation, and competing ions and such information and would assist in evaluating the mobility and therefore the bioavailability of AsV at contaminated sites. The objectives of this study were to examine the kinetics of AsV adsorption and desorption in the presence of different ions that have been reported to influence AsV adsorption.
Materials and methods
Soil
The Ah horizon (0–100 mm) of an uncontaminated soil was collected from an uncultivated site in northern New South Wales, Australia. This soil represents one of the major soil groups in this region, and the area has a well-documented history of As contamination through the application of As-based pesticides for the control of cattle ticks. Many of the soils surrounding the former cattle dip sites contain elevated As concentrations ranging between <100 to >14,000 mg kg−1 (Smith et al. 2003). The soil sample collected from the field was air-dried and crushed to pass through a 2-mm stainless-steel sieve. Following crushing, the soil was stored in an airtight polythene container for analyses. Pertinent properties of the soils are shown in Table 1, and analyses of these properties are as described previously by Smith et al. (1999).
Kinetic experiments
Apparatus
The stir-flow reaction chamber used was a modification of the stir-flow chamber developed by Carski and Sparks (1985). It was constructed from stainless steel and consisted of three parts (Fig. 1): (1) a reaction chamber 22.5 mm in diameter with a maximum depth of 40 mm, (2) an adjustable plunger with an inlet port through the centre of the plunger and (3) a cover with a teflon seal and an outlet port. Over the outlet port, a 25-mm diameter 0.45-μm nylon filter was fitted to retain soil in the reaction chamber. The position of the inlet port in the stirred-flow chamber differed from that developed by Carski and Sparks (1985) as it was observed in preliminary studies that the mixing of the soil by the magnetic stirrer led to a significant transfer of soil from the mixing chamber to the inlet port. Modification of the stirred-flow cell reaction chamber included the fitting and repositioning of a filter to the inlet port.

Design of stirred-flow reactor modified from Carski and Sparks (1985)
A magnetic stirrer was used to ensure adequate mixing in the reaction chamber. The stirring speed was maintained at a rate that ensured good mixing of soil particles, but with minimum soil particle abrasion. The speed of the magnetic stirrer was standardised for all experiments. A flow rate of 1.5 ml min−1 was kept constant throughout the experiment, with both the inlet and outlet solutions being pumped using a peristaltic pump.
Experimental
Adsorption kinetics
A soil sample (0.5 g) and the teflon-coated magnetic stirrer were placed in the reaction chamber. To this, 5 ml of 27 μM AsV solution in a background electrolyte was quickly added to the reaction chamber, and the chamber was immediately sealed. The same solution that was added to the reaction chamber was then pumped into the reaction chamber at a rate of 1.5 ml min−l. This flow rate was periodically checked throughout the studies. Effluent solution was collected with a fraction collector at various sample periods, ranging between 0.25 to 10 min. All experiments were conducted at a temperature of 20 ± 2°C.
Initially, blank background curves for AsV sorption without soil in the chamber were determined with the same AsV solution that was used in later experiments. The kinetic experiments were conducted in duplicate for both the blank (background) and AsV sorption curves. Time zero was defined as the moment of entry of the first drop of effluent solution into the first tube of the fraction collector. The As concentration in the effluent solution was analysed using flame atomic absorption spectrometry with hydride generation, and the amount of AsV adsorbed was calculated from the difference between the amount added and amount remaining in solution. Solution pH and other ions also present in the effluent solution were also analysed where sufficient solution was collected by inductively coupled plasma atomic emission spectroscopy.
The effect of ionic strength (I) on the kinetics of AsV adsorption was studied in either (1) 0.03 M NaNO3 or (2) 0.3 M of NaNO3 in the presence of 27 μM AsV in each of the background electrolytes. The effect of different index cations was studied at the same ionic strength using 0.03 M of NaNO3 and 0.01 M of Ca(NO3)2. Anion competition was studied with the sorption studies conducted in the presence of phosphate (P). Phosphate (32 μM P) was added in solution with 27 μM AsV in a background electrolyte of either 0.03 M NaNO3 or 0.01 M Ca(NO3)2. The concentrations of AsV and P utilised in this study were based on an analysis of extractable (soil:water 1:2) AsV and P concentrations in soil solution at 12 former cattle dip sites in northern New South Wales, Australia.
Desorption kinetics
Initially soil (0.5 g) was equilibrated with AsV (27 μM AsV) in a background solution of 0.03 M NaNO3 as described above for 2 h. Adsorption studies had shown that AsV adsorption was rapid and essentially completed within 2 h with a maximum amount of 200 μg g−1 AsV sorbed. Desorption was then initiated by passing through the chamber solutions containing (1) 0.03 M NaNO3, (2) 0.3 M NaNO3 or (3) 0.03 M NaNO3 and 32 μM P.
Blank curves for AsV desorption without soil in the chamber were determined using solutions with the same composition. Analyses of AsV in effluent solutions were conducted as described above. Duplicate experiments were conducted for all desorption curves.
Data analysis
The cumulative amount of AsV adsorbed by the soil can be calculated from flow rates and concentrations of AsV in the effluent solutions (Schnabel and Fitting 1988). Arsenate adsorbed with time was evaluated using Eq. 1 (Yin et al. 1997), where q a (t i ) is the cumulative sorption at time t i (μg g−1), C i is the effluent concentration for the ith sampling period (μg l−1), J is the flow rate (l min−1), Δt i is the sampling time interval (min), c(t i ) is the concentration in the chamber at t i (μg l−1), V is the volume of the reaction chamber (l), W is the soil concentration in the chamber (g l−1), and subscripts s and b refer to the chamber with and without soil present, respectively.
The first term on the right-hand side of the Eq. 1 contains experimental data, while the second term is calculated for specific times (Eick et al. 1990). The calculation can be done exactly if the kinetic model is known (Schnabel and Fitting 1988) or it can be approximated by averaging the effluent concentration for that time period and the succeeding one (Yin et al. 1997). The second method was used in this study. Estimated values are close to the real values when short time intervals are used (Schnabel and Richie 1987).
The cumulative amount of AsV desorbed is given by Eq. 2;
where q d (t i ) is the cumulative AsV desorbed at time t i (μg g−1).
The use of Eqs. 1 and 2 to calculate the cumulative amount of AsV adsorbed and desorbed by soil accounts for both the effect of dilution and the amount of AsV adsorbed by the stirred-flow reaction chamber (Yin et al. 1997). Several assumptions are made when studying sorption processes by the stirred-flow method. These have been highlighted by Yin et al. (1997) and are reiterated here. It is assumed that the sorption kinetics of AsV by the stirred-flow chamber is not affected by the presence of soil in the chamber. Although this is improbable, the presence of soil is likely to decrease the sorption rates of AsV by the chamber. Furthermore, the presence of soil will affect the rate of AsV adsorbed by the stirred-flow chamber for only a very short period of time as the stirred-flow reaction chamber adsorbs only a small amount of AsV compared with the amount of AsV adsorbed by the soil.
The kinetic data were fitted to a number of mathematical equations, including first order, second order, simple Elovich, and parabolic diffusion equations. The applicability of such empirical equations has been explained extensively by Sparks (1989). However, for the purpose of these studies, it should not be implied from the fitting of data to one particular kinetic equation that this is the only equation that could be used to describe the apparent sorption coefficients. Apparent sorption coefficients were determined so those trends in different treatments could be assessed. Data were also fitted to a one-site second-order model (Eq. 3) to determine the apparent rate coefficients (Yin et al. 1997);
where q a is the concentration of AsV adsorbed by soil at time t (μg g−1), k a is the forward rate coefficient (l μg−1 min−1), q ∞ is the concentration of AsV adsorbed at equilibrium (μg g−1), C t is the AsV solution concentration in the reaction chamber at time t (μg l−1), k −a is the reverse rate coefficient (min−1), and subscripts t and Δt indicate quantities of time (min).
The desorption reaction of AsV from the soil can be expressed as:
where q o is the desorbable AsV concentration on the soil at the beginning, q d is the concentration of AsV desorbed from the soil at time t (μg g−1), k d is the desorption rate coefficient (min−1), and k −d is the reverse rate coefficient (l μg−1 min−1).
The model used by Yin et al. (1997) assumed that there is only one type of site involved in the sorption reaction. Although this represents a simplistic view of the heterogenous nature of the soil, it allows some insight into the reaction mechanisms between adsorbent and adsorbate. Yin et al. (1997) showed that for HgII the reverse reactions were insignificant, and the Eqs. 3 and 4 simplified to Eqs. 5 and 6.
Therefore, Eqs. 5 and 6 were used to describe the experimental data from these studies.
Statistical analysis
All statistical analyses were conducted using GENSTAT 5 (1997). The parameters for Eqs. 5 and 6 (k a, k d, q ∞ and q o) were determined for the soil treatments by fitting a non-linear regression model that optimised the function in Eqs. 5 and 6. The graphical fit of the model, together with the residual diagnostics and standard errors of the parameter estimates, was investigated for assessing the model fit to the experimental data.
Initial statistical analyses suggested that the residuals from the non-linear regression modelling showed evidence of lagged correlation with time (i.e., values for a particular time are dependent on previous time point values). As a consequence, attention is not focussed on R 2 values, but rather it is placed on the graphical fit of the model.
The parameter estimates for the unknown functions in Eqs. 5 and 6 (k a, k d, q ∞ and q o) were computed via two sets of analyses. Firstly, where more than one experimental run was undertaken for each treatment, the values were averaged and then the models fitted. The other analysis obtained parameter estimates for each run of the treatments, and these estimates were used to determine whether there were treatment differences between the parameters. In addition to this, data were modelled for varying lengths of experimental runs.
Results and discussion
Sorption kinetics
Preliminary studies were conducted before studying the adsorption–desorption kinetics of AsV to ascertain whether AsV-soil reactions were instantaneous or kinetically controlled (Bar-Tal et al. 1990). If non-equilibrium conditions exist in the reaction chamber then stopping the flow through the chamber for a sufficient period of time before equilibrium conditions are established will result in a noticeable drop in the concentration of effluent solution once the flow is restarted. However, if the reaction is instantaneous then a plot of the effluent concentration versus time should be continuous. In the preliminary study, stopping the flow after 5 min for approximately 25 min reduced the effluent concentration of AsV by 24% (data not shown), indicating that non-equilibrium conditions exist within the reaction chamber and AsV sorption by the Alfisol is time dependent. Therefore, kinetic studies utilising the stirred-flow reactor are applicable for the sorption of AsV.
The kinetics of AsV sorption by the Alfisol with different treatments are shown in Fig. 2. The sorption of AsV was initially fast (58–91% in the first 15 min), and this was followed by sorption at a slower rate that continued with all treatments for the duration of the study period. This biphasic sorption pattern is similar to HgII sorption by different soils observed by Yin et al. (1997) and has been reported in many other sorption studies in the literature. Fuller et al. (1993) and Raven et al. (1998) have reported similar AsV sorption kinetic behaviour on ferrihydrite surfaces, although Raven et al. (1998) reported that the rate of AsV sorption was dependent on solution pH and the AsV solution concentration. Similarly, Grafe and Sparks (2005) reported that the rate of AsV sorption by goethite was dependent on a range of factors including the number of sorption sites. Fendorf et al. (1997) have reported that at low AsV surface coverage of an Fe oxide surface, AsV is mainly retained through the formation of monodentate complexes and bidentate binuclear and bidentate mononuclear complexes at high AsV surface coverage. Raven et al. (1998) have postulated that the formation of bidentate complexes may be slower than the formation of monodentate complexes. Another possibility is that diffusion controlled reactions into soil particles may be responsible for the slow AsV sorption phase observed. Fuller et al. (1993) suggested that the slow sorption of AsV may be due to diffusion controlled reactions into the ferrihydrite crystal structure and modelled the kinetics using the parabolic diffusion equation, which has been used previously to model kinetic reactions in soils (Aharoni et al. 1991). In this study, the parabolic diffusion equation did not usually describe the data well (data not shown). However, this does not exclude the possibility that diffusion processes influence the slow reaction phase of AsV sorption. Aharoni et al. (1991) observed that several mathematical expressions may describe the sorption kinetics, and the sorption processes probably involve more than one sorption mechanism. Therefore, one mathematical equation may not adequately describe the sorption of AsV observed in this study.

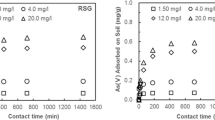
Effect of different treatments on the cumulative amount of AsV adsorbed by the Alfisol soil. Bars represent standard errors of means. Where no bar is seen, error is smaller than the symbol
In some experiments, AsV sorption was not observed to apparent equilibrium. However, using GENSTAT 5 (1997) it was possible to predict the cumulative sorption capacity of the soil (q ∞) and optimise the other unknown functions in Eq. 5. It is therefore possible to compare the effect of the different treatments on the sorption capacity of AsV by the Alfisol soil. The sorption capacities of the soil without the presence of P in the influent solution were similar (Table 2) irrespective of the nature of the index cation and I of the soil solution. However, the presence of P decreases the estimated sorption capacity from 162 to 89 and 177 to 115 μg g−1 AsV in 0.03 M NaNO3 and 0.01 M Ca(NO3)2, respectively. Studies examining the effect of anion or cation-induced adsorption have reported several mechanisms may enhance the adsorption of the counter-ion at the surface. In the case of Ca or Cd adsorption in the presence of P, for example, the mechanisms may include: (1) precipitation of Cd2+ as Cd3(PO4)2, (2) co-adsorption of HPO4 2− and Cd2+ as an ion pair, (3) surface complex formation of Cd2+ onto the adsorbed HPO4 2− and (4) HPO4 2− induced Cd2+ adsorption (Bolan et al. 1999). Sadusky and Sparks (1991) observed that the amount of potassium (K) adsorbed by two soils (coarse-loamy, siliceous, thermic Typic Hapludult and loamy, siliceous, thermic Arenic Hapludult) at pH 5 and 6 was dependent on the accompanying anion. Sorption of K in the presence of anions, such as those of silicate (Si) and P, was greater than with chloride (Cl). This was attributed to the change in surface charge imparted when Si and P are specifically adsorbed onto variable-charge surfaces. Hundal and Pasricha (1998) studied the effect of temperature on the sorption of K by a soil (Udic Ustochrept) and reported that the cumulative amount of K adsorbed decreased with increasing temperature. They also observed that K sorption was greater in the presence of Cl compared with perchlorate (ClO4). The authors attributed the increase in sorption of K in the presence of chloride to the formation of weak Ca–Cl complexes that decrease Ca competition with K for sorption sites on soil.
The presence of P has generally been reported to decrease AsV sorption when both anions are present in solution (Roy et al. 1986; Manning and Goldberg 1996; Darland and Innskeep 1997). Parfitt (1978) suggested that the sorption order of anions on soil surfaces was phosphate > arsenate > selenite = molybdate > sulphate = fluoride > chloride > nitrate. In this study, sorption sites are continually being exposed to a constant concentration of P and AsV due to the addition and removal of solution from the stirred-flow reaction chamber. With the continual replacement of solution in the stirred-flow reaction chamber, it is observed that P only decreases AsV adsorbed by between 35 to 48%. If the binding energy is the major factor controlling sorption of P or AsV, then at similar solution concentrations, P adsorbed will always be greater than AsV irrespective of the number of available sorption sites. The effect of P on AsV sorption may be indicative of the presence of sites that are specific for the sorption of either AsV or P. It was hypothesised that increasing the number of available sorption sites would increase the amount of AsV adsorbed and may affect the rate of the sorption process. Increasing the number of sorption sites through the addition of 1% goethite with the Alfisol soil resulted in an increase in the amount of AsV adsorbed (data not shown). No kinetic data were obtainable after 15 min due to clogging of the stirred-flow reaction chamber. However, the kinetic data collected clearly illustrate that the number of available sorption sites strongly influences the cumulative amount and rate of AsV adsorbed by the soil.
Several kinetic equations were fitted to describe the apparent rate of AsV sorption on soil so that the effects of different treatments could be compared (Table 3). The various kinetic equations tested all had limitations in describing the apparent rate of AsV sorption by soil. This is evidenced by the fact that the first-order equation, which has often been used to describe reaction rates, described the sorption of AsV by soil well for the Na treatments, but not for the Ca treatments. This may be indicative of a different sorption mechanism influencing the sorption of AsV in the presence of Ca. As discussed by Smith et al. (2002), the presence of Ca in solution may increase AsV sorption in low adsorbing soils through an increase in the surface positive charge of the soil surface. However, the nature of these sorption sites is unknown, and the AsV mechanism may not be the same as in the presence of other index cations such as Na.
For all the treatments, the simple Elovich equation was found to describe AsV sorption reasonably well (data not shown). The Elovich equation assumes a heterogeneous distribution of sorption energies, and the energy of activation increases linearly with surface coverage (Schnabel and Fitting 1988; Sparks 1989). Due to the nature of soil, there is a continuum of sorption sites that have a range of different sorption energies, and therefore, it would be expected that an empirical equation based on this mechanism may be applicable to soil. Although the Elovich equation has been widely used to describe kinetic data (Chien and Clayton 1980; Sparks and Jardine 1984), Ungarish and Aharoni (1981) have questioned the value of using the Elovich equation to describe the adsorption and desorption kinetics at very low and very high surface coverage due to the “poor fit” of data to the equation. This was also observed in this study (data not shown), with a “poor fit” of the collected data to the Elovich equation after short (low surface coverage) and long (high surface coverage) sampling times. It was therefore decided to use the one-site second-order equation (Eq. 5) rather than the Elovich equation to determine the apparent forward rate constants of the reactions.
The AsV sorption data fitted Eq. 5 well, and this is reflected in the low values of SE and RMS for the estimated parameters (Table 3). This implies that although the nature of the adsorbing surface differs, the assumption that there is one type of sorption site may be consistent at high surface coverage of AsV on the adsorbing surfaces. This is supported to some extent by Fendorf et al. (1997) who postulated that AsV formed three different types of surface structure on goethite depending on the surface coverage. Monodentate complexes were postulated to form at low surface coverage and bidentate complexes at high surface coverage, but a mixture of the two complexes may also occur at surface coverage in between. However, further studies need to be conducted to substantiate the kinetic model of sorption proposed by Yin et al. (1997).
The calculated average k a values for all but the goethite treatments are shown in Table 3. The goethite treatment is not included because of the lack of experimental data. There were differences between the estimated k a values for the different treatments, although an analysis of variance (GENSTAT 5 1997) showed that the differences were significant only at p = 0.068. Notably, the k a values for the 0.03 M NaNO3 and the 0.03 M NaNO3 plus P treatments appear to be different k a values compared to the other treatments. This may indicate that these treatments had different effects on the rate of AsV adsorbed. However, a comparison of similar treatments [0.01 M Ca(NO3)2 compared with 0.03 M NaNO3, 0.01 M Ca(NO3)2 + P compared with 0.03 M NaNO3 + P] indicates the sorption capacity of the soil is similar in each treatment, although the rate of the sorption reaction is different. Investigations of the sorption kinetics of other ions have shown that k a may or may not be affected by different soil treatments. Sadusky and Sparks (1991) reported that the nature of the anion and increasing soil pH from 5 to 6 had little effect on the apparent rate of K sorption by soils, although the amount of K adsorbed differed greatly with different anions and pH. In contrast, Toner et al. (1989) reported that increasing the pH of three soils (Typic Hapludult and Plinthic Paleudult) from pH 5 to 7 decreased the cumulative amount of nitrate (NO3 −) sorbed, but increased the rate of NO3 − sorbed. Toner et al. (1989) attributed the observed increase in k a to fewer sorption sites available for NO3 − sorption, due to the increasing pH of the soil surface, which increases the overall rate of sorption. Generally, there is little or no information available in the literature regarding the kinetics (rate and mechanisms) affecting AsV sorption. Although AsV sorption has been well studied, much of this research has been conducted after an equilibrium period has been attained. Using equilibrium methods is tenuous as the behaviour of contaminants in natural systems is almost always in disequilibrium. Further studies investigating these disequilibrium reactions are needed as the initial sorption reactions play a critical role in the subsequent sorption behaviour of AsV (Grafe and Sparks 2005).
Desorption kinetics
Considerably less AsV was desorbed than was adsorbed (Fig. 3). At the end of 100 min of desorption, only 2, 6 and 17% of the adsorbed AsV was desorbed from the Alfisol soil in the presence of 0.3 M NaNO3, 0.03 M NaNO3 and 0.03 M NaNO3 + P, respectively. This suggests that once adsorbed, much of the adsorbed AsV cannot be readily desorbed into solution. The persistence of adsorbed metals in soils has often been reported (Bruemmer et al. 1988; Yin et al. 1997; McLaren et al. 1998) and has been attributed to diffusion into mineral lattices (Sparks et al. 1980) or through intraparticle pores (Sparks et al. 1980), but as noted earlier, the parabolic diffusion equation did not describe the data well.

Effect of different treatments on the cumulative amount of AsV desorbed from the Alfisol soil. Bars represent standard errors of means. Where no bar is seen, error is smaller than the symbol. Total amount of As adsorbed prior to desorption was 200 μg g−1
The specific sorption of AsV to high affinity sites in soil may contribute to the observed slow reversibility of AsV. Extended edge X-ray absorption studies by Waychunas et al. (1993) and Fendorf et al. (1997) reported that AsV formed inner-sphere monodentate and bidentate bonds on the surface of Fe oxides, and the preferential bonding to high affinity sites may contribute to the slow reversibility. Although P was observed to increase the amount of AsV desorbed compared with the other treatments (Fig. 3), increasing the influent P concentration to 160 μM after 100 min of desorption in the presence of 32 μM P only resulted in a small increase in the amount of AsV desorbed (result not shown). This may indicate that some AsV remains irreversibly bound to the soil irrespective of the soil treatment. Darland and Innskeep (1997) have reported similar findings after studying the desorption of AsV from aquifer packed column material contaminated with AsV. They reported that even in the presence of P concentrations that exceeded the sorption capacity of the column by two orders of magnitude, approximately 40% of sorbed AsV remained bound to the aquifer materials.
Increasing I from 0.03 to 0.3 M NaNO3 resulted in a smaller increase in the cumulative amount of AsV desorbed compared with the amount of AsV desorbed in the presence of P. Analysis of variance (GENSTAT 5 1997) showed that there was a significant difference (p < 0.05) in the amounts of AsV desorbed for the different treatments. The increase in the cumulative amount of AsV desorbed in the presence of P compared with I may be related to the nature of the mechanisms by which P and I influence the AsV adsorbed by soil surfaces. Phosphate may compete with AsV for surface sorption sites when sites are limited (Manning and Goldberg 1996; Smith et al. 1999), and therefore direct competition between AsV and P may result in the increased desorption of AsV. Changing I, however, may influence AsV adsorbed only through its effects on the surface potential of the colloid (Smith et al. 1999). Therefore, once AsV is adsorbed, changes to the surface potential of the adsorbing colloid had little influence on the amount of AsV desorbed.
The small values of SE and RMS (Table 4) indicated that the one-site second order equation (Eq. 6) described the desorption data well. Although the three desorption treatments had different effects on the amount of AsV desorbed, an analysis of variance (GENSTAT 5 1997) found that there was little difference between k d values (Table 4) with differences in the k d values only observed at p = 0.44. It could be hypothesised that irrespective of the total amount of AsV desorbed by different treatments, the k d of AsV desorption from the Alfisol does not alter. However, this hypothesis is drawn from fitting of the data to the one-site second order equation (Eq. 6) and is not drawn on any scientific observation. The equation does not describe how the different ions interact on the soil surface, and further studies are needed in this area.
Conclusions
The sorption of AsV on the Alfisol soil was characterised by an initially rapid phase, followed by a slower phase. The different treatments had little effect on the total amount of AsV adsorbed by the Alfisol, as was observed in the presence of different cations or varying the ionic strength of the background solution, although the rate at which the maximum adsorption was achieved varied depending on the treatment. In contrast, the presence of P decreased the amount of AsV adsorbed by the Alfisol significantly. Although the apparent rate of AsV sorption varied with sorption treatments, an analysis of variance showed that the differences were significant only at p = 0.068. The amount of AsV desorbed from the Alfisol represented less than 17% of the AsV adsorbed. There were differences in the total amount desorbed for different treatments, with the presence of P greatly increasing the amount of AsV desorbed. These results suggest that AsV adsorption kinetics play an important role in controlling the transport and bioavailability of AsV in the soil environment.
References
Aharoni, C., Levison, S., Ravina, I., & Sparks, D. L. (1991). Kinetics of soil chemical reactions: Relationships between empirical equations and diffusion models. Soil Science Society of America Journal, 55, 1307–1312.
Amacher, M. C. (1991). Methods of obtaining and analysing kinetic data. In D. L. Sparks & D. L. Suarez (Eds.), Rates of chemical processes. Madison, USA: Soil Science Society of America.
Anderson, M. A., Ferguson, J. F., & Gavis, J. (1976). Arsenate adsorption on amorphous aluminium hydroxide. Journal of Colloid and Interface Science, 54, 391–399.
Bar-Tal, A., Eick, M. J., Feigenbaum, S., Sparks, D. L., & Fishman, S. (1995). Determination of rate coefficients for potassium–calcium exchange on vermiculite using a stirred-flow chamber. Soil Science Society of America Journal, 59, 760–765.
Bar-Tal, A., Sparks, D. L., Pesek, J. D., & Feigenbaum, S. (1990). Analyses of adsorption kinetics using a stirred-flow chamber: I. Theory and critical tests. Soil Science Society of America Journal, 54, 1248–1253.
Bolan, N. S., Naidu, R., Syers, J. K., & Tillman, R. W. (1999). Surface charge and solute interaction in soils. Advances in Agronomy, 67, 88–141.
Bruemmer, G. W., Gerth, J., & Tiller, K. G. (1988). Reaction kinetics of the adsorption and desorption of nickel, zinc and cadmium by goethite. I. Adsorption and desorption of metals. Journal of Soil Science, 39, 37–52.
Carski, T. H., & Sparks, D. L. (1985). A modified miscible displacement technique for investigating adsorption–desorption kinetics in soils. Soil Science Society of America Journal, 49, 1114–1116.
Chien, S. H., & Clayton, W. R. (1980). Application of the Elovich equation to the kinetics of phosphate release and sorption in soils. Soil Science Society of America Journal, 44, 265–268.
Darland, J. E., & Innskeep, W. P. (1997). Effects of pH and phosphate competition on the transport of arsenate. Journal of Environment Quality, 26, 1133–1139.
Eick, M. J., Bar-Tal, A., Sparks, D. L., & Feigenbaum, S. (1990). Analyses of adsorption kinetics using a stirred-flow chamber. Soil Science Society of America Journal, 54, 1278–1282.
Elkhatib, E. A., Bennett, O. L., & Wright, R. J. (1984). Arsenite sorption and desorption in soils. Soil Science Society of America Journal, 48, 1025–1030.
Fendorf, S., Eick, M. J., Grossl, P., & Sparks, D. L. (1997). Arsenate and chromate retention mechanisms on goethite. 1. Surface structure. Environmental Science and Technology, 31, 315–320.
Fuller, C. C., Davis, J. A., & Waychunas, G. A. (1993). Surface chemistry of ferrihydrite: Part 1. EXAFS studies of the geometry of coprecipitated and adsorbed arsenate. Geochimica et Cosmochimica Acta, 57, 2271–2282.
GENSTAT 5. (1997). Release 4.1. Lawes agricultural trust (3rd ed.). Rothamsted, England: ICAR.
Grafe, M., & Sparks, D. L. (2005). Kinetics of zinc and arsenate co-sorption at the goethite–water interface. Geochimica et Cosmochimica Acta, 69, 4573–4595.
Harter, R. D. (1991). Kinetic of sorption/desorption processes in soil. In D. L. Sparks & D. L. Suarez (Eds.), Rates of chemical processes. Madison, USA: Soil Science Society of America.
Hundal, L. S., & Pasricha, N. S. (1998). Adsorption–desorption kinetics of potassium as influenced by temperature and background anions. Geoderma, 83, 215–225.
Manning, B. A., & Goldberg, S. (1996). Modeling competitive adsorption of arsenate with phosphate and molybdate on oxide minerals. Soil Science Society of America Journal, 33, 121–131.
Manning, B. A., & Goldberg, S. (1997a). Adsorption and stability of arsenic(III) at the clay mineral–water interface. Environmental Science and Technology, 31, 2005–2011.
Manning, B. A., & Goldberg, S. (1997b). Arsenic(III) and arsenic(V) adsorption on three California soils. Soil Science, 162, 886–895.
McLaren, R., Naidu, R., Smith, J., & Tiller, K. G. (1998). Fractionation and distribution of arsenic in soils contaminated by cattle dip. Journal of Environment Quality, 27, 348–354.
Parfitt, R. L. (1978). Anion adsorption by soils and materials. Advances in Agronomy, 30, 1–50.
Pierce, M. L., & Moore, C. B. (1980). Adsorption of arsenite and arsenate on amorphous iron hydroxide from dilute aqueous solutions. Environmental Science and Technology, 14, 214–216.
Raven, K. P., Jain, A., & Loeppert, R. H. (1998). Arsenite and arsenate adsorption on ferrihydrite: Kinetics, equilibrium and adsorption envelopes. Environmental Science and Technology, 32, 344–349.
Roy, W. R., Hassett, J. J., & Griffin, R. A. (1986). Competitive interactions of phosphate and molbdate on arsenate sorption. Soil Science, 142, 203–210.
Sadusky, M. C., & Sparks, D. L. (1991). Anionic effects on potassium reactions in variable-charge Atlantic coastal plain soils. Soil Science Society of America Journal, 55, 371–375.
Schnabel, R. R., & Fitting, D. J. (1988). Analyses of chemical kinetics data from dilute, dispersed, well-mixed flow-through systems. Soil Science Society of America Journal, 52, 1270–1273.
Schnabel, R. R., & Richie, E. B. (1987). Elimination of the time assignment bias in estimates of dispersion coefficient. Soil Science Society of America Journal, 51, 302–304.
Scott, M. J., & Morgan, J. J. (1995). Reactions of oxide surfaces. I. Oxidation of As(III) by synthetic birnessite. Environmental Science and Technology, 29, 1898–1905.
Seyfried, M. S., Sparks, D. L., Bar-Tal, A., & Feigenbaum, S. (1989). Kinetics of calcium–magnesium exchange on soil using a stirred-flow reaction chamber. Soil Science Society of America Journal, 53, 406–410.
Skopp, J. (1986). Analysis of time-dependent chemical processes in soils. Journal of Environment Quality, 15, 205–213.
Smith, E., Naidu, R., & Alston, A. M. (1999). Sorption of arsenate and arsenite by four Australian soils. Journal of Environment Quality, 28, 1719–1726.
Smith, E., Naidu, R., & Alston, A. M. (2002). Chemistry of inorganic arsenic in soils: II. Effect of P, Na and Ca. Journal of Environment Quality, 31, 557–563.
Smith, E., Smith, J., Smith, L., Biswas, T., Correll, R., & Naidu, R. (2003). Arsenic in the Australian environment: An overview. Journal of Environmental Science and Health Part A, 38, 223–239.
Sparks, D. L. (1989). Kinetics of soil processes. New York: Academic Press.
Sparks, D. L., & Jardine, P. M. (1984). Comparison of kinetic equations to describe potassium–calcium exchange in pure and mixed systems. Soil Science, 138, 115–122.
Sparks, D. L., Zelazny, L. W., & Martens, D. C. (1980). Kinetics of potassium exchange in a paleudult from the coastal plain of Virginia. Soil Science Society of America Journal, 44, 37–40.
Toner, C. V., I. V., Sparks, D. L., & Carski, T. H. (1989). Anion exchange chemistry of middle Atlantic soils: Charge properties and nitrate retention kinetics. Soil Science Society of America Journal, 53, 1061–1067.
Ungarish, M., & Aharoni, C. (1981). Kinetics of chemisorption: Deducing kinetic laws from experimental data. Journal of Chemical Society Faraday Transactions, 1, 975–979.
Waychunas, G. A., Rea, B. A., Fuller, C. C., & Davis, J. A. (1993). Surface chemistry of ferrihydrite: Part 1. EXAFS studies of the geometry of coprecipitated and adsorbed arsenate. Geochimica et Cosmochimica Acta, 57, 2251–2269.
Yin, Y., Allen, H. E., Huang, C. P., Sparks, D. L., & Sanders, P. F. (1997). Kinetics of mercury(II) adsorption and desorption on soil. Environmental Science and Technology, 31, 496–503.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Smith, E., Naidu, R. Chemistry of inorganic arsenic in soils: kinetics of arsenic adsorption–desorption. Environ Geochem Health 31 (Suppl 1), 49–59 (2009). https://doi.org/10.1007/s10653-008-9228-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10653-008-9228-z