
Abstract
Pharmaceuticals are increasingly found in aquatic ecosystems due to the non-efficiency of waste water treatment plants. Therefore, aquatic organisms are frequently exposed to a broad diversity of pharmaceuticals. Freshwater snail Radix balthica has been chosen as model to study the effects of oxazepam (psychotropic drug) on developmental stages ranging from trochophore to hatching. In order to provide a global insight of these effects, a transcriptome deep sequencing has been performed on exposed embryos. Eighteen libraries were sequenced, six libraries for three conditions: control, exposed to the lowest oxazepam concentration with a phenotypic effect (delayed hatching) (TA) and exposed to oxazepam concentration found in freshwater (TB). A total of 39,759,772 filtered raw reads were assembled into 56,435 contigs having a mean length of 1579.68 bp and mean depth of 378.96 reads. 44.91% of the contigs have at least one annotation. The differential expression analysis between the control condition and the two exposure conditions revealed 146 contigs differentially expressed of which 144 for TA and two for TB. 34.0% were annotated with biological function. There were four mainly impacted processes: two cellular signalling systems (Notch and JNK) and two biosynthesis pathways (Polyamine and Catecholamine pathways). This work reports a large-scale analysis of differentially transcribed genes of R. balthica exposed to oxazepam during egg development until hatching. In addition, these results enriched the de novo database of potential ecotoxicological models.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Aquatic organisms are frequently exposed to a great diversity of chemicals suspended in water or adsorbed onto substrates. Among these compounds, pharmaceuticals are a class of emerging environmental contaminants increasingly found in aquatic ecosystems. These pollutants are released in the environment mainly via Waste Water Treatment Plant (WWTP) effluents. Several studies have demonstrated that pharmaceuticals are not completely removed by WWTP, their elimination being dependent on the specific treatment methods used (Calisto and Esteves 2009; Yuan et al. 2013) but also on their abundance in the waste water, their pharmacokinetics, and their physical and chemical properties (Loos et al. 2013; Chen et al. 2015). Among pharmaceuticals, psychiatric drugs are the most frequently detected in aquatic environments (Fong and Ford 2014). Among the molecules, anxiolytics are a frequently prescribed class in which benzodiazepines such as oxazepam and carbamazepine are the most commonly used (Brodin et al. 2013). They are frequently detected in WWTP effluents and surface water. As an example, they have been found in 27 to 100% of French freshwaters (Idder et al. 2013; Bouissou-Schurtz et al. 2014) and their concentrations are ranged from 161 ng.L−1 (Bouissou-Schurtz et al. 2014) to 816 ng.L−1 (Chiffre et al. 2016). The presence of these compounds may have a short and/or long term (depending on the compounds release and persistence characteristics) impact on freshwater biodiversity (Sanchez et al. 2011).
In studies on aquatic ecosystems, molluscs, including snails, are commonly used to evaluate pollutant effects (Cheung and Lam 1998; Coeurdassier et al. 2003; Byrne et al. 2009; Das and Khangarot 2010; Coutellec et al. 2013). Our study used the pond snail Radix balthica (Linnaeus 1758, Basommatophora, Pulmonata), as an experimental model since it is present in many French rivers and European water bodies (Seddon et al. 2013). We studied the embryonic and larval stages because of their greater sensitivity to contaminants compared to the adult snails (Gomot 1998). Several works have documented the phenotypic impact of contaminant exposure on Lymnaea such as Lymnaea stagnalis. Some of these studies involved monitoring targeted biomarkers such as activity of the detoxification enzymes or expression of their encoding genes (e.g., cytochrome P450 (CYP450), glutathione-S-transferase (GST)) to assess the biological effects of pollutants (Bouétard et al. 2013; Gust et al. 2013; Bonnafé et al. 2015). They revealed an impact on one or more enzymes of detoxification system, while impacts on other networked functions were not demonstrated. It is also becoming necessary to use approaches which provide wider views of these effects. Interesting works have focused on genes expression of different aquatic organisms in responses to a pollutant or environmental changes (Wang et al. 2009; Bouétard et al. 2012). All these global transcriptomic works shown many impacted genes revealing potential key functions perturbations. Although very few analyses are available to establish a pattern of impacted genes or functions. The reproducibility of such studies is also challenging since the methods used for bioinformatics treatments have not been normalized and most studies apply different in-house command lines. Otherwise, the access to a reference genome of model organism defines the power and the depth of the analyses. Despite the absence of a sequenced genome that decreases depth and power of the analyses, the transcriptomic studies can yield precious information’s on transcriptome sequences for further research such as targeting RT-PCR or even in cladistics. Radix balthica presents the advantage of spawning fertilised eggs in a gelatinous mass giving access to complete embryonic development, from the two-cell stage to the hatchling (Sarker et al. 2007; Pande 2010).
In this current work, we characterized the transcriptomic profiles of freshly hatched snails previously exposed to oxazepam solution during their embryonic development. Rather than focusing on well-studied biomarkers and molecular pathways associated to drug responses, our aim was to identify new candidates by comparing transcriptome profiles of snails exposed to oxazepam solution at 0.815 and 10 µg.L−1 to snails in a control condition. The dose of 0.815 µg.L−1 represent the surface water concentration of oxazepam as previously described in the PSYCHEAU project manuscript 2014 (Unpublished data), and the higher one is the lowest concentration for which a phenotypic effect was previously observed on Japanese medaka Oryzias latipes larvae (Chiffre et al. 2016). The transcriptome profiles were generated by RNA-sequencing with an Illumina HiSeqTM 2000 sequencing system and were then compared to highlight the differentially expressed (DE) genes in each condition. This study gave us the global impact of oxazepam on the R. balthica transcriptome.
Materials and methods
Animals and oxazepam exposure experiment
The experimental R. balthica were obtained from the River Caussel (Albi, France, latitude: 43.931157; longitude: 2.167686) in June 2014 and acclimated for 1 week prior to collecting the egg masses. They were reared in a 40 L tank with aerated reconstituted AFNOR water (Gomot 1998) (NaHCO3 200 mg.L−1; CaCl2.2H2O 297 mg.L−1; MgCl2.6H2O 167 mg.L−1; anhydrous K2SO4 26 mg.L−1) and river freshwater (v/v) at a temperature of 20 ± 1 °C. During the acclimation period, the snails were fed with organic lettuce ad libitum under day/night photoperiod of 14/10 h.
The oxazepam used for the experiment was purchased from Alsachim (Strasbourg, France). A minimal concentration of ethanol (0.001%) was used to facilitate powder dissolution. One control condition with 0.001% ethanol supplemented AFNOR water and two concentrations, 10 µg.L−1 (TA) and 0.815 µg.L−1 (TB), the effective concentration and the environmental concentration respectively, were used for RNA extraction and sequencing. After 1 week of acclimation, egg masses laid were collected, and only the trochophore stage selected. Five egg masses were dissected to form a single egg pool. To follow embryonic development, each egg was placed into a different well of a 96 well plate. Eggs were randomly distributed into three experimental groups (control, TA and TB) with 90 individuals per group.
Sampling and total RNA extraction
Six replicates of ten neonate snails (not older than 24 h) for each condition (control, treated TA, treated TB) were used for RNA extraction and sequencing. The snails were recovered randomly after hatching, pooled per condition and homogenized with 200 µL TRIzol® Reagent (Ambion™ by Life Technologies, France).
Total RNA was extracted following the TRIzol® manufacturer’s instructions. Total RNA was cleaned using RNeasy® Micro Kit (Qiagen, France) following the instructions of RNeasy Micro Handbook (12/2007, p56) with on column DNase I treatment and then stored at −80 °C. RNA purity and quantity was checked using the NanoDrop™ 2000 (Thermo Scientific, France) and RNA quality was determined with Agilent RNA 6000 Nano kit of Agilent 2100 Bioanalyzer (Agilent Technologies). The automated and standardized method for RNA quality control provided by the software relies on calculation of an RNA integrity number (RIN). The RIN calculation employs numerous features, including 18S and 28S fragment regions height, area, and intercept of the baseline (Schroeder et al. 2006).
In mollusc, 28S RNA fragment is rarely observed. As documented in a range of invertebrates (Ishikawa 1977; Barcia et al. 1997; Muttray et al. 2008; Winnebeck et al. 2010), 28S absence is due to a breaking point in the rRNA structure which converts 28S into 2 fragments that are hydrogen bounded and that migrate approximately at the same size as the 18S rRNA during gel electrophoresis. Therefore we visually assessed the total RNA quality by evaluating the absence of oligonucleotides (produced by RNA degradation) and the presence of the peak corresponding to both rRNA.
Library preparation and RNA sequencing
The 18 libraries were prepared from 1.1 µg of total RNA with the TruSeq® Stranded mRNA LT Kit (Illumina®) following the manufacturer’s recommendations and then checked with the High Sensitivity chip of the Agilent 2100 BioAnalyzer (Agilent Technologies). The libraries were then quantified in qPCR with the ABI 7900HT (Applied Biosystems). Library preparations were sequenced with TruSeq® PE Cluster v3 kit (Illumina®), TruSeq® SBS 200 cycle v3 kit (Illumina®) and TruSeq® Multiplex Primer kit (Illumina®) on Hiseq 2000 (Illumina®) with generation of paired-end (2 × 100pb) reads.
De novo transcriptome assembly and annotation
Per condition assemblies
First the read pairs were stored, the database quality checking and the cleaning of remaining sequencing adapters were done using trim_galore (available at http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/, version0.4.0) (Mariette et al. 2012). Then the over-represented reads were filtered out using the normalize_by_kmer_coverage.pl command lines from the Trinity software package (Grabherr et al. 2011). The next step aimed at discarding the invalid base calls by extracting the longest sub-sequence without Ns from each read. If the length of the longest sub-sequence did not exceed half of the sequence length, the read and its mate were removed. This last step was performed using an in-house command lines.
Nine Oases assemblies using nine different k-mers (25, 31, 37, 43, 49, 55, 61, 65, and 69) were performed on the pre-processed input data (Schulz et al. 2012). Each assembly produces a transcripts.fa file. Each raw_transcripts.fa file is then organized into loci. Only the longest and most covered (used reads) loci are kept using the OasesV0.2.04OutputToCsvDataBase.py command lines developed by a Brown University team. After that, all files were merged. Finally, anti-sense chimeras (accidentally produced by the assembly step) were split with a homemade script.
Because similar collections of contigs are produced by different k-mers, a cd-hit-est step was used to remove contigs based on their sequence similarities (Fu et al. 2012). Because different kmers sometimes construct different parts of the transcripts, we used TGICL to assemble contigs having significant overlaps (Pertea et al. 2003). The contigs were also filtered on a minimum length of 200 base pairs.
Input reads were then mapped back to the contigs using bwa aln (version 0.7.6) (Li and Durbin 2009). The resulting alignment files were used to correct the contig sequences from spurious insertions and deletions using an in-house command lines and to filter out those with very low coverage. The filter excludes contigs with less than two mapped reads per million.
Meta-assembly
All single condition contig fasta files were concatenated and each contig renamed by adding the condition name at the beginning of the contig name. The longest Open Reading Frame (ORF) was then searched with getorf from EMBOSS (Rice et al. 2000). Cd-hit clustering was performed on the ORFs with sequence identity equal to or greater than 90% in order to extract, from each cluster, the contig with the longest ORF or, if the ORF size was the same, the longest contig. Clustering using cd-hit-est was performed on the remaining contigs with sequence identity equal to or greater than 95%. Input reads from all conditions were then mapped to the contigs using bwa aln (Li and Durbin 2009). Contigs with very low coverage, less than 2 mapped reads per million, were filtered out.
Variant discovery
The raw reads were realigned to the final set of contigs using bwa mem (Li and Durbin 2009). The alignments were then filtered on alignment unicity and quality using the Q30 threshold. The resulting bam files were re-aligned and recalibrated using GATK version 2.4.9 (McKenna et al. 2010). SNP and INDEL calling was performed using the Unified genotyper algorithm of GATK. SSR were searched in the contig fasta file using trf (Benson 1999).
Differential expression analysis
The differential expression detection of genes across samples was performed using the DESeq R package (1.14.0) (Anders and Huber 2012). Briefly, the analysis was performed after the estimation of effective library size, also called normalization, with “estimateSizeFactor” function and after variance estimation with the “estimateDispersions” function.
The DE contigs between control and TA, control and TB, TA and TB were identified with “nbinomTest” resulting in a data frame with p-values that were fitted using Benjamini and Hochberg’s procedure. An adjusted p value of 0.05 was used as the threshold to significantly select the DE contigs. Among the significantly DE contigs, we classified them according to their over (Fold Change > 1) or under-expressed (Fold Change < 1) profile.
Contig annotation and function prediction
The contigs were aligned vs. different nucleic and protein reference databases including: SwissProt, RefSeq protein, Limnea stagnalis contig, Crassostrea gigas and Lottia gigantea Ensembl protein, Biomphalaria glabrata peptides. The GO terms were retrieved using the SwissProt, RefSeq protein alignment matches. All the results were stored in an RNAbrowse (Mariette et al. 2014) instance.
Function prediction was determined using Interproscan which assigned Gene Ontology (GO) terms for contigs having at least one significant hit against an annotated reference from a database. For differentially expressed contigs, GO annotation was mapped into three ontologies (“Biological Process”, “Molecular Function” and “Cellular Component”) using Generic GO Term Mapper and then plotted using the WEGO (Web Gene Ontology Annotation Plot) plotting tool.
Functional annotation of DE contigs was performed using KAAS (Kyoto Encyclopedia of Gene and Genome (KEGG) Automatic Annotation Server) and KOBAS (KEGG Orthology Based Annotation System) 2.0 to test the statistical enrichment of DE genes in the KEGG pathway. Only pathways with a corrected p value < 0.05 were considered as significantly enriched. Results of annotation were then categorized using KEGG pathways, BRITE hierarchies for protein family analysis and KEGG modules.
Sequence accession numbers
The original sequencing reads of R. balthica juveniles libraries were submitted on European Nucleotide Archive (ENA) and are available under accession number PRJEB12711 (http://www.ebi.ac.uk/ena/data/view/PRJEB12711).
Results
Analysis of libraries and contig assemblies
The 18 libraries generated a total of 503,171,850 raw reads before filtering with the Illumina CASAVA 1.8 filter which deleted 39,759,772 raw reads (7.90% of the total). Then, the filtered reads were cleaned before assembling. A total of 56,435 contigs was generated (Table 1). Mean contig length and mean contig depth were calculated using all contigs from the 18 libraries. They were respectively 1579.68 bp (Fig. 1) and 378.96 reads (Table 1).

Contig distribution according to length. The contigs length was calculated using all contigs from the 18 libraries
Analysis of mapping
Reads were mapped to the contig assembly to measure the “gene” expression level for each contig. For each of the eighteen libraries, an average of 97% clean reads were mapped to a contig in the assembled contig data (Fig. 2).

Clean reads mapping to assembled contigs for each library. Control refers to control condition, Oxa_10 refers to 10 µg.L−1 Oxazepam condition and Oxa_0.815 refers to 0.815 µg.L−1 oxazepam condition
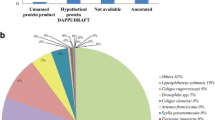
Annotation
The BLAST search annotated 44.91% of the 56,435 contigs (Fig. 3): 24.55% against RefSeqProt, 7.85% against the L. stagnalis contig database, 5.54% against the B. glabrata peptide database, 3.57% against the L. gigantea peptide database, 1.94% against the C. gigas peptide database and 1.46% against SwissProt.

Graphical representation of contig annotation
Differential expression
The DESeq package used for the DE contigs revealed 144 DE contigs, with 59 contigs up-regulated and 85 down-regulated for treatment A vs. control. For treatment B vs. control, only 2 contigs appeared to be up-regulated. Further analysis showed that only 34.0% of the DE contigs were annotated with a biological function (Table 2, see supplementary Table S1 for complete data). For TA, among the most annotated under-expressed contigs, there was function prediction of Morc (Members of the conserved Microrchidia family) family protein, Dentin sialophosphoprotein (DSPP), Carbonyl reductase and also proteins of ADAM (A Disintegrin And Metalloprotease) family, with fold change range from 0.058 to 0.328. Among the lowest ones, there was function prediction of enzymes such as Beta-galactosidase protein, Calumenin protein, Ribonuclease and L-Fucosidase proteins with fold change range from 0.728 to 0.745. For the over-expressed annotated contigs, the lowest impacted had function predictions of Ganglioside activator, Tyrosine receptor protein and Puromycin sensitive aminopeptidase with fold change range from 1.454 to 1.507 and the most over-expressed ones had function predictions of Hemagglutinin protein, Mitogen activated protein kinase (MAPK) and Deleted in malignant brain tumours protein with fold change range from 2.236 to 3.101. For TB, the only annotated contig was over-expressed and predicted as Otopetrin protein with fold change of 1.644.
Functional annotation analysis of DE contigs
All the available GO-terms of the 144 DE contigs of TA and the 2 DE contigs of TB were mapped and plotted into three ontologies (Fig. 4) using WEGO plot. The most enriched ontology was “molecular function” with four dominant functions, “binding”, “catalytic”, “hydrolase” and “ion binding”.

Distribution of the over and under-expressed contigs using GO terms according to three GO top categories. C refers to control condition, TA refers to 10 µg.L−1 oxazepam exposure
KOBAS 2.0 software was used to identify the most significantly enriched KEGG pathway which is represented in Table 3. The 2 DE contigs of the TB are not found in any of the pathways. The 6 most enriched pathways of the DE contigs of TA were the “Notch signalling pathway” with 5 contigs assimilated to this pathway, “Chemical carcinogenesis” with 2 contigs assimilated to this pathway, “Arachidonic acid metabolism” enriched with 2 contigs, “Epithelial cell signalling in Helicobacter pylori” with 2 contigs assimilated for this pathway, “Extracellular matrix receptor (ECM-receptor) interaction” with 2 contigs assimilated to this pathway and “Aminobenzoate degradation” enriched with 1 contig (Table 3). The KEGG Mapper was used for BRITE hierarchy reconstruction (Fig. 5), which classifies the K numbers of each contig into three ontologies named “Metabolism”, “Genetic Information Processing” and “Signalling and Cellular Processes”. The most represented ontology was “Metabolism” with the Enzyme subcategory containing 15 contigs. The KEGG Mapper was also used for module reconstruction of the 144 differentially expressed contigs of the TA, the 2 DE contigs of TB haven’t highlighted a module. That resulted in 6 modules representing the main principal modules affected by differentially expressed contigs. One contig (Rp1_contig_28236) encodes a predicted Mitogen Activated Protein Kinase Kinase Kinase 2 (MAP3K2 protein), potentially involved in three modules (three MAPK pathway), c-Jun N-terminal kinase also called JNK (M00688), p38 (M00689) and Extracellular signal-Regulated Kinase also called ERK (M00690). Another module was Notch signalling (M00682) with 5 contigs down-regulated (Rp1_LOC101849541.1.7, Rp1_LOC101849541.2.7, Rp1_LOC101849541.7.7, Rp1_LOC100533548 and Rp1_LOC102304721.1.2). The alignment of the three contigs encoding a predicted ADAM 17 protein (Rp1_LOC101849541.1.7, Rp1_LOC101849541.2.7, Rp1_LOC101849541.7.7) lead us to conclude to an assembling error as these contigs appeared to be parts of the same sequence encoding a predicted ADAM 17 protein. The two other contigs, Rp1_LOC100533548 and Rp1_LOC102304721.1.2, were predicted to encode ADAM 10 and Deltex protein, respectively. The last two modules impacted were Catecholamine biosynthesis (M00042) with one contig up-regulated (Rp1_contig_36516) encoding a predicted function of Dopamine β–hydroxylase (DBH) and Polyamine biosynthesis (M00133) with one down-regulated contig (Rp1_contig_38845) encoding a predicted Agmatinase (AGMAT) protein.

Graphical representation of differentially expressed contigs according to the KEGG top categories. The BRITE hierarchy reconstruction was based on the K numbers of each differentially expressed contig under TA (10 µg.L−1 oxazepam) exposure
Insertion, deletion (INDEL) and single nucleotide polymorphism (SNP) detection
The detection of INDELs and SNPs performed with the GATK algorithm identified 455,826 SNPs (92.4% of variants) and 37,517 INDELs (7.6% of variants) with 19,628 insertion (4% of variants) and 17,889 deletion (3.6% of variants).
Discussion
In this study, RNA sequencing produced 56,435 contigs with a mean length of 1579.68 bp which increases probability to find a complete Open Reading Frame (ORF) in one contig and thus facilitates prediction of functional annotation. Thus, 44.91% present an annotated ORF. A higher mean length must be problematic because of the possibility to find two or more ORFs in one contig which can disrupt DE analysis. In the present case, the mean contig length was suitable to yield an almost complete transcript.
Differential analysis
Differential expression analysis revealed 144 contigs DE for treatment A. Only 34.0% of contigs were annotated with various functions such as enzyme activity, cell surface receptors or proteins involved in cell signalling processes. The other contigs could be orphan proteins, non-coding RNA or sequences from UTR protein regions. Moreover, we can’t discard the presence of partially or misassembled transcript, because of the absence of genome sequencing data and functional annotations available for non-model organisms (Schunter et al. 2014). For treatment B, only 2 contigs were DE and only one was annotated as Otopetrine. Several hypotheses can explain the lack of contigs DE under this condition. First, the few number of sequenced genomes limits the annotation power and sometimes may generates two or more partial or complete Open Reading Frame (ORF) on one contig. Schunter et al. (2014) has demonstrated that numbers of contig isoforms increase with contig length resulting in a substantially reduction of differential analysis efficiency. This point being dependant on the in-house command lines used, could also affect the annotation. This bias decreases severely the assembling and the annotation strength, particularly for the DE contigs treatment. In addition, the method used for bioinformatics treatment could be too stringent (to delete the false positive). Some genes didn’t appear DE while they are not, in some case, data structures need to accommodate multiple transcripts per locus due to alternative splicing and sequences that are repeated in different genes introduce ambiguity (Grabherr et al. 2011). Another hypothesis would be that effect of low concentrations of oxazepam were difficult to detect on the transcriptome of R. balthica. The impact could be visible at another level of the organism such as proteome or enzyme activities level. As shown by Chen et al. (2014), the low concentration of pharmaceuticals like carbamazepine (at 0.5 µg/L) used in this study impact catalase activity (higher activity of catalase in the digestive gland and gills of Corbicula fluminea exposed for 30 days to carbamazepine solution) but had no effect on genes expression of Heat shock protein (Hsp). These observations support the hypothesis that low concentrations of oxazepam could impact significantly another biological mechanism. These limited effects could be also due to the too long or too short exposure time tested.
After function prediction of the 144 DE contigs of treatment A made with the GO database, molecular function appears to be the most enriched ontology with “Catalytic” and “Hydrolase” function being the most represented. This observation is in line with the Brite hierarchy reconstruction of the KEGG database using K-numbers, with the “Metabolism” ontology being the most represented with “Enzyme” sub-categorie. Function prediction using the KEGG database highlighted two major impacted pathways: “Notch signalling pathway” involved in many cellular processes and “Extra Cellular Matrix receptor (ECM-receptor) interaction” involved mainly in cell-cell interactions (Table 3). KEGG Mapper prediction emphasizes the presence among the DE contigs of “Notch” module but also of the “MAPK signalling” (JNK) module and two biosynthesis pathways i.e. “Catecholamine” and “Polyamine” biosynthesis. The potential disturbance of these four predicted pathways is discussed below.
Notch signalling pathway
As reviewed by Weinmaster et al. (Weinmaster et al. 1992; Weinmaster 1997, 1998, 2000) the Notch signalling pathway is a complex cascade of events conserved in many eukaryotic cells which culminates in altered genes expression patterns. Notch signal transduction is mainly initiated by cell-cell contact and triggers a succession of reactions involving many partners such as metalloprotease enzyme (ADAM proteins family) or ubiquitin transferase enzyme (Deltex protein). Our study highlighted down-regulation of three contigs encoding two predicted ADAM proteins family (ADAM 10 and ADAM 17) and one predicted Deltex (DTX) protein. The ADAM protein family mediates the second cleavage of mature cell surface receptor required for the third cleavage of Notch that releases the Notch active cytoplasmic domain from the plasma membrane. ADAM 17 (also called TACE) is also involved in many other pathways with a critical role in the processing of multiple protein like Tumor Necrosis Factor (TNF) proteins family (Peschon et al. 1998) and interestingly it has been shown that it could be inhibited by benzodiazepine molecules in mammalian organisms like mouse (Nelson et al. 2002; Levin et al. 2004; Scheller et al. 2011; Dreymueller et al. 2015). Our results are in accordance with the studies of Levin et al. (2004), Nelson et al. (2002) and Scheller et al. (2011), which showed TACE inhibition by many of substituted benzodiazepine hydroxamates compounds. In our case, accepting the hypothesis that Notch signalling operate in the same way of mammalian signal transduction, the inhibition of metalloprotease enzyme could decrease the active cytoplasmic form of Notch and so decrease the transcription of Notch target genes. Notch proteins could be directly inhibited by benzodiazepine molecules. As shown by Gavai et al. (2015), the benzodiazepine compound like 2-oxo-1,4-benzodiazepin-3-yl-succinamides inhibits in vitro the γ-secretase mediated signalling of Notch receptors. The role of Deltex protein is not clear in the Notch pathway but recent studies suggest that Deltex protein interacts with other proteins in the cytoplasm and in the nucleus to activate or inhibit the transcription of target genes (Diederich et al. 1994; Matsuno et al. 1995; Weinmaster 1997; Baron 2012). Deltex encoding gene is also a target of Notch signalling (Deftos et al. 1998). The down-regulation of the deltex gene in our study could be the result of Notch inhibition via the down-regulation of the ADAM genes. The scenario where Notch, ADAM 10, ADAM 17 would be inhibited could potentially have an impact on many mechanisms, such as neurogenesis, myogenesis, but also cellular specification, differentiation, proliferation and survival. One or more of these mechanisms occurring during embryogenesis could be impacted by oxazepam exposure and more broadly by benzodiazepines. This disturbance could have a strong teratogenic impact on our model and thus on the fitness of organisms exposed in the environment.
Mitogen-activated protein kinases
The second module which appeared to be impacted was the mitogen-activated protein kinases (MAPK). Three MAP kinase cascades have been characterised: ERK, JNK and p38. Our study highlighted the over-expression of one contig encoding predicted MAP3K2 serine/threonine protein kinase mainly involved in the MAP-JNK signalling pathway. As reviewed by Takeda and Ichijo (2002), the JNK signalling pathway consists of three classes of serine/threonine kinases, MAPK, MAPK Kinase (MAPKK or MEK) and MAPKK kinase (MAPKKK). MAP3K2 is the first serine/threonine kinase phosphorylated in the JNK pathway. It is known that the JNK pathway is mainly activated in response to environmental stress such as UV, heat shock or osmotic shock and also by proinflammatory cytokines and interleukin-1 (Tumor Necrosis Factor and ILK-1) (Takeda and Ichijo 2002). Interestingly, it has been shown that Notch and Nuclear factor-kappa B (NF-κB) pathways can inhibit the MAP-JNK pathway via the Growth arrest and DNA-damage-inducible (Gadd45) protein (Elsharkawy et al. 2005; Maniati et al. 2011). This observation supports the impact on Notch signalling pathway suggested by our results. Only one contig over-expressed represented the MAPK module predicted via KEGG module reconstruct. The other partners of MAP3K2 may be over-expressed but undetected as differentially expressed because of the stringent bioinformatics methods used for differential expression analysis.
Catecholamine biosynthesis pathway
CAs are a class of neurotransmitters mainly represented by dopamine (DA) and its metabolic products, noradrenaline (NA) and adrenaline (A). These three CAs are synthesized from the amino-acid L-tyrosine using four principal enzymes. Our study showed one over-expressed contig encoding a predicted Dopamine-β-hydroxylase (DBH) protein involved in DA hydroxylation which produces NA (Flatmark 2000). As shown by Richetto et al. (2015) on the mouse model organism, the positive allosteric modulation of the GABA receptor α-5 subunit may be particularly useful in mitigating pathological overactivity of the dopaminergic system. In our study, we used oxazepam, an agonist of the GABA receptor. The over-expression of DBH suggests possible over-production of dopamine neurotransmitter which could also affect the noradrenaline and adrenaline synthesis pathways. This suggestion is in accordance with results published by Heikkinen et al. (2008). They have shown a modulation of the glutamatergic transmission mediated by benzodiazepines which activates the ventral tegmental area (VTA) dopamine neurons and increases the amount of dopamine released in the nucleus accumbens (associated to pleasure, incentive salience, etc.), increasing the benzodiazepines impacts. Even if only one contig appeared DE in this module, it is interesting to consider these results because this neurotransmitter is involved in many physiological processes, such as cell-cell nervous conduction, but also in movement, feeding and behaviour, very important physiological processes for population fitness.
Polyamine biosynthesis pathway
The conventional polyamine biosynthesis pathway from arginine to polyamine is mediated by Arginase and Ornithine decarboxylase (ODC1) proteins. Some studies showed an alternative to this conventional pathway using Arginine decarboxylase (ADC) and Agmatinase (AGMAT) (Halaris and Plietz 2007; Wang et al. 2014). Our study highlighted the inhibition of one contig encoding predicted AGMAT protein involved in another polyamine pathway from arginine to putrescine via ADC and AGMAT protein (Halaris and Plietz 2007; Wang et al. 2014). It seems that these two pathways have a complementary role. This suggests a possible decrease in the amount of polyamines such as putrescine, spermine and spermidine which could lead to disturbance of DNA and protein synthesis regulation, the scavengers of reactive oxygen species, cell proliferation and the differentiation of tissues. The inhibition of AGMAT would also trigger an increase of Agmatine protein (precursor of putrescine) amount and thus disturb many cellular processes which involve Agmatine such as voltage gated Ca2+ channels and consequently nervous conduction of adrenergic, serotoninergic or also cholinergic neurones (Reis and Regunathan 2000). Agmatine can also modulates the release of glutamate and noradrenaline (Halaris and Plietz 2007). Thereby, the modulation of agmatine production could directly impact glutamatergic and noradrenergic nervous conduction. Although this is based on a single contig in the module, it is consistent with our results suggesting that benzodiazepine could disrupt the noradrenaline and dopamine nervous conduction.
Conclusion
This study revealed a strong impact on the R. balthica transcriptome after exposure to 10 µg.L−1 oxazepam. Interestingly, at environmental concentration, no detectable effects were observed on Radix embryo transcriptome. These disturbances on the expression patterns of R. balthica may impact on important functions related to the homeostasis of development in this freshwater snail. Moreover, this study allowed enrichment of non-model organism transcript database used in ecotoxicological approaches.
References
Anders S, Huber W (2012) Differential expression of RNA-Seq data at the gene level–the DESeq package. Heidelberg, Germany: European Molecular Biology Laboratory (EMBL)
Barcia R, Lopez-García JM, Ramos-Martínez JI (1997) The 28S fraction of rRNA in molluscs displays electrophoretic behaviour different from that of mammal cells. Biochem Mol Biol Int 42:1089–1092
Baron M (2012) Endocytic routes to Notch activation. Semin Cell Dev Biol 23:437–442. doi:10.1016/j.semcdb.2012.01.008
Benson G (1999) Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27:573–580
Bonnafé E, Sroda S, Budzinski H et al. (2015) Responses of cytochrome P450, GST, and MXR in the mollusk Corbicula fluminea to the exposure to hospital wastewater effluents. Environ Sci Pollut Res 22:11033–11046. doi:10.1007/s11356-015-4309-x
Bouétard A, Besnard A-L, Vassaux D et al. (2013) Impact of the redox-cycling herbicide diquat on transcript expression and antioxidant enzymatic activities of the freshwater snail Lymnaea stagnalis. Aquat Toxicol 126:256–265. doi:10.1016/j.aquatox.2012.11.013
Bouétard A, Noirot C, Besnard A-L et al. (2012) Pyrosequencing-based transcriptomic resources in the pond snail Lymnaea stagnalis, with a focus on genes involved in molecular response to diquat-induced stress. Ecotoxicology 21:2222–2234. doi:10.1007/s10646-012-0977-1
Bouissou-Schurtz C, Houeto P, Guerbet M et al. (2014) Ecological risk assessment of the presence of pharmaceutical residues in a French national water survey. Regul Toxicol Pharmacol 69:296–303. doi:10.1016/j.yrtph.2014.04.006
Brodin T, Fick J, Jonsson M, Klaminder J (2013) Dilute concentrations of a psychiatric drug alter behavior of fish from natural populations. Science 339:814–815. doi:10.1126/science.1226850
Byrne RA, Rundle SD, Smirthwaite JJ, Spicer JI (2009) Embryonic rotational behaviour in the pond snail Lymnaea stagnalis: influences of environmental oxygen and development stage. Zoology 112:471–477. doi:10.1016/j.zool.2009.03.001
Calisto V, Esteves VI (2009) Psychiatric pharmaceuticals in the environment. Chemosphere 77:1257–1274. doi:10.1016/j.chemosphere.2009.09.021
Chen H, Zha J, Liang X et al. (2014) Effects of the human antiepileptic drug carbamazepine on the behavior, biomarkers, and heat shock proteins in the Asian clam Corbicula fluminea. Aquat Toxicol 155:1–8. doi:10.1016/j.aquatox.2014.06.001
Chen M, Cooper VI, Deng J et al. (2015) Occurrence of pharmaceuticals in Calgary’s wastewater and related surface water. Water Environ Res 87:414–424. doi:10.2175/106143015X14212658614199
Cheung CCC, Lam PKS (1998) Effect of cadmium on the embryos and juveniles of a tropical freshwater snail, Physa acuta (Draparnaud, 1805). Water Sci Technol 38:263–270
Chiffre A, Clérandeau C, Dwoinikoff C, et al. (2016) Psychotropic drugs in mixture alter swimming behaviour of Japanese medaka (Oryzias latipes) larvae above environmental concentrations. Environ Sci Pollut Res 23(6):4964–4977. doi:10.1007/s11356-014-3477-4
Coeurdassier M, De Vaufleury A, Badot P-M (2003) Bioconcentration of cadmium and toxic effects on life-history traits of pond snails (Lymnaea palustris and Lymnaea stagnalis) in laboratory bioassays. Arch Environ Contam Toxicol 45:102–109. doi:10.1007/s00244-002-0152-4
Coutellec M-A, Besnard A-L, Caquet T (2013) Population genetics of Lymnaea stagnalis experimentally exposed to cocktails of pesticides. Ecotoxicology 22:879–888. doi:10.1007/s10646-013-1082-9
Das S, Khangarot BS (2010) Bioaccumulation and toxic effects of cadmium on feeding and growth of an Indian pond snail Lymnaea luteola L. under laboratory conditions. J Hazard Mater 182:763–770. doi:10.1016/j.jhazmat.2010.06.100
Deftos ML, He Y-W, Ojala EW, Bevan MJ (1998) Correlating notch signaling with thymocyte maturation. Immunity 9:777–786
Diederich RJ, Matsuno K, Hing H, Artavanis-Tsakonas S (1994) Cytosolic interaction between deltex and Notch ankyrin repeats implicates deltex in the Notch signaling pathway. Dev Camb Engl 120:473–481
Dreymueller D, Uhlig S, Ludwig A (2015) ADAM-family metalloproteinases in lung inflammation: potential therapeutic targets. Am J Physiol Lung Cell Mol Physiol 308:L325–343. doi:10.1152/ajplung.00294.2014
Elsharkawy AM, Oakley F, Mann DA (2005) The role and regulation of hepatic stellate cell apoptosis in reversal of liver fibrosis. Apoptosis 10:927–939. doi:10.1007/s10495-005-1055-4
Flatmark (2000) Catecholamine biosynthesis and physiological regulation in neuroendocrine cells. Acta Physiol Scand 168:1–17. doi:10.1046/j.1365-201x.2000.00596.x
Fong PP, Ford AT (2014) The biological effects of antidepressants on the molluscs and crustaceans: A review. Aquat Environ 151:4–13. doi:10.1016/j.aquatox.2013.12.003
Fu L, Niu B, Zhu Z et al. (2012) CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28:3150–3152. doi:10.1093/bioinformatics/bts565
Gavai AV, Quesnelle C, Norris D et al. (2015) Discovery of clinical candidate BMS-906024: a potent pan-notch inhibitor for the treatment of leukemia and solid tumors. . ACS Med Chem Lett 6:523–527. doi:10.1021/acsmedchemlett.5b00001
Gomot A (1998) Toxic effects of cadmium on reproduction, development, and hatching in the freshwater snail Lymnaea stagnalis for water quality monitoring. Ecotoxicol Environ Saf 41:288–297. doi:10.1006/eesa.1998.1711
Grabherr MG, Haas BJ, Yassour M et al. (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29:644–652. doi:10.1038/nbt.1883
Gust M, Fortier M, Garric J et al. (2013) Effects of short-term exposure to environmentally relevant concentrations of different pharmaceutical mixtures on the immune response of the pond snail Lymnaea stagnalis. Sci Total Environ 445–446:210–218. doi:10.1016/j.scitotenv.2012.12.057
Halaris A, Plietz J (2007) Agmatine: metabolic pathway and spectrum of activity in brain. CNS Drugs 21:885–900. doi:10.2165/00023210-200721110-00002
Heikkinen AE, Möykkynen TP, Korpi ER (2008) Long-lasting modulation of glutamatergic transmission in VTA dopamine neurons after a single dose of benzodiazepine agonists. Neuropsychopharmacology 34:290–298. doi:10.1038/npp.2008.89
Idder S, Ley L, Mazellier P, Budzinski H (2013) Quantitative on-line preconcentration-liquid chromatography coupled with tandem mass spectrometry method for the determination of pharmaceutical compounds in water. Anal Chim Acta 805:107–115. doi:10.1016/j.aca.2013.10.041
Ishikawa H (1977) Comparative studies on the thermal stability of animal ribosomal RNA’s V. Tentaculata (phoronids, moss-animals and lamp-shells). Comp Biochem Phys B 57:9–14. doi:10.1016/0305-0491(77)90073-6
Levin JI, Nelson FC, Delos Santos E et al. (2004) Benzodiazepine inhibitors of the MMPs and TACE. Part 2. Bioorg Med Chem Lett 14:4147–4151. doi:10.1016/j.bmcl.2004.06.031
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. doi:10.1093/bioinformatics/btp324
Loos R, Carvalho R, António DC et al. (2013) EU-wide monitoring survey on emerging polar organic contaminants in wastewater treatment plant effluents. Water Res 47:6475–6487. doi:10.1016/j.watres.2013.08.024
Maniati E, Bossard M, Cook N et al. (2011) Crosstalk between the canonical NF-κB and Notch signaling pathways inhibits Pparγ expression and promotes pancreatic cancer progression in mice. J Clin Invest 121:4685–4699. doi:10.1172/JCI45797
Mariette J, Escudié F, Allias N et al. (2012) NG6: Integrated next generation sequencing storage and processing environment. BMC Genomics 13:462. doi:10.1186/1471-2164-13-462
Mariette J, Noirot C, Nabihoudine I et al. (2014) RNAbrowse: RNA-seq de novo assembly results browser. PLoS One 9:e96821. doi:10.1371/journal.pone.0096821
Matsuno K, Diederich RJ, Go MJ et al. (1995) Deltex acts as a positive regulator of Notch signaling through interactions with the Notch ankyrin repeats. Dev Camb Engl 121:2633–2644
McKenna A, Hanna M, Banks E et al. (2010) The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303. doi:10.1101/gr.107524.110
Muttray AF, Schulte PM, Baldwin SA (2008) Invertebrate p53-like mRNA isoforms are differentially expressed in mussel haemic neoplasia. Mar Environ Res 66:412–421. doi:10.1016/j.marenvres.2008.06.004
Nelson FC, Delos Santos E, Levin JI et al. (2002) Benzodiazepine inhibitors of the MMPs and TACE. Bioorg Med Chem Lett 12:2867–2870
Pande GS (2010) Observations on the embryonic development of freshwater pulmonate snail Lymnaea acuminata (lamarck, 1822) (gastropoda: mollusca). The Bioscan 5:549–554
Pertea G, Huang X, Liang F et al. (2003) TIGR gene indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics 19:651–652. doi:10.1093/bioinformatics/btg034
Peschon JJ, Slack JL, Reddy P et al. (1998) An essential role for ectodomain shedding in mammalian development. Science 282:1281–1284
Reis DJ, Regunathan S (2000) Is agmatine a novel neurotransmitter in brain?. Trends Pharmacol Sci 21:187–193. doi:10.1016/S0165-6147(00)01460-7
Rice P, Longden I, Bleasby A (2000) EMBOSS: the European molecular biology open software suite. Trends Genet TIG 16:276–277
Richetto J, Labouesse MA, Poe MM et al. (2015) Behavioral effects of the benzodiazepine-positive allosteric modulator SH-053-2’F-S-CH3 in an Immune-mediated neurodevelopmental disruption model. Int J Neuropsychopharmacol 18:pyu055–pyu055. doi:10.1093/ijnp/pyu055
Sanchez W, Sremski W, Piccini B et al. (2011) Adverse effects in wild fish living downstream from pharmaceutical manufacture discharges. Environ Int 37:1342–1348. doi:10.1016/j.envint.2011.06.002
Sarker MM, Nesa B, Jahan MS (2007) Embryonic developmental ecology of freshwater snail Lymnaea acuminata (Lymnaeidae: Gastropoda). Pak J Biol Sci PJBS 10:23–31
Scheller J, Chalaris A, Garbers C, Rose-John S (2011) ADAM17: a molecular switch to control inflammation and tissue regeneration. Trends Immunol 32:380–387. doi:10.1016/j.it.2011.05.005
Schroeder A, Mueller O, Stocker S et al. (2006) The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Mol Biol 7:3. doi:10.1186/1471-2199-7-3
Schulz MH, Zerbino DR, Vingron M, Birney E (2012) Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 28:1086–1092. doi:10.1093/bioinformatics/bts094
Schunter C, Vollmer SV, Macpherson E, Pascual M (2014) Transcriptome analyses and differential gene expression in a non-model fish species with alternative mating tactics. BMC Genomics 15:167. doi:10.1186/1471-2164-15-167
Seddon MB, Kebapçi U, Van Damme D, Prie V (2013) Radix balthica. In: IUCN red list threat. Species. http://www.iucnredlist.org/details/155647/0. Accessed 11 Jun 2015
Takeda K, Ichijo H (2002) Neuronal p38 MAPK signalling: an emerging regulator of cell fate and function in the nervous system. Genes Cells 7:1099–1111. doi:10.1046/j.1365-2443.2002.00591.x
Wang X, Ying W, Dunlap KA et al. (2014) Arginine decarboxylase and agmatinase: an alternative pathway for de novo biosynthesis of polyamines for development of mammalian conceptuses. Biol Reprod 90:84–84. doi:10.1095/biolreprod.113.114637
Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10:57–63. doi:10.1038/nrg2484
Weinmaster G (1997) The Ins and outs of notch signaling. Mol Cell Neurosci 9:91–102. doi:10.1006/mcne.1997.0612
Weinmaster G (1998) Notch signaling: direct or what?. Curr Opin Genet Dev 8:436–442. doi:10.1016/S0959-437X(98)80115-9
Weinmaster G (2000) Notch signal transduction: a real Rip and more. Curr Opin Genet Dev 10:363–369. doi:10.1016/S0959-437X(00)00097-6
Weinmaster G, Roberts V, Lemke G (1992) Notch2: a second mammalian Notch gene. Development 116:931–941
Winnebeck EC, Millar CD, Warman GR (2010) Why does insect RNA look degraded?. J Insect Sci 10:1–7. doi:10.1673/031.010.14119
Yuan S, Jiang X, Xia X et al. (2013) Detection, occurrence and fate of 22 psychiatric pharmaceuticals in psychiatric hospital and municipal wastewater treatment plants in Beijing, China. Chemosphere 90:2520–2525. doi:10.1016/j.chemosphere.2012.10.089
Acknowledgements
The authors thank Olivier Bouchez and all Get-PlaGe operators, INRA of Castanet-Tolosan for the perfect processing of our unusual samples. Funding for this work was provided to J-YM by a grant from the Region Midi-Pyrénées, Toulouse (France) for 70% and from Jean-François Champollion University Institute, Albi (France) for 30%. We thank also Jean-Michel MALGOUYRES operator for his help in organism collection and in maintain rearing of R. balthica.
Authors’ contributions
The project was conceived and planned by FG, EB and J-YM. J-YM exposed, grinded and extracted the RNA. CK and FE performed the bioinformatics analysis and J-YM performed the differential analysis and the annotation of differential expressed contigs. J-YM wrote the manuscript and FG and EB proofread it.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval
All procedures performed in this study were in accordance with the ethical standards of the institution at which the study was conducted and was in compliance with current French laws.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Mazzitelli, JY., Bonnafe, E., Klopp, C. et al. De novo transcriptome sequencing and analysis of freshwater snail (Radix balthica) to discover genes and pathways affected by exposure to oxazepam. Ecotoxicology 26, 127–140 (2017). https://doi.org/10.1007/s10646-016-1748-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10646-016-1748-1