
Abstract
Despite the knowledge on anaerobic degradation of hydrocarbons and signature metabolites in the oil reservoirs, little is known about the functioning microbes and the related biochemical pathways involved, especially about the methanogenic communities. In the present study, a methanogenic consortium enriched from high-temperature oil reservoir production water and incubated at 55 °C with a mixture of long chain n-alkanes (C15–C20) as the sole carbon and energy sources was characterized. Biodegradation of n-alkanes was observed as methane production in the alkanes-amended methanogenic enrichment reached 141.47 μmol above the controls after 749 days of incubation, corresponding to 17 % of the theoretical total. GC–MS analysis confirmed the presence of putative downstream metabolites probably from the anaerobic biodegradation of n-alkanes and indicating an incomplete conversion of the n-alkanes to methane. Enrichment cultures taken at different incubation times were subjected to microbial community analysis. Both 16S rRNA gene clone libraries and DGGE profiles showed that alkanes-degrading community was dynamic during incubation. The dominant bacterial species in the enrichment cultures were affiliated with Firmicutes members clustering with thermophilic syntrophic bacteria of the genera Moorella sp. and Gelria sp. Other represented within the bacterial community were members of the Leptospiraceae, Thermodesulfobiaceae, Thermotogaceae, Chloroflexi, Bacteroidetes and Candidate Division OP1. The archaeal community was predominantly represented by members of the phyla Crenarchaeota and Euryarchaeota. Corresponding sequences within the Euryarchaeota were associated with methanogens clustering with orders Methanomicrobiales, Methanosarcinales and Methanobacteriales. On the other hand, PCR amplification for detection of functional genes encoding the alkylsuccinate synthase α-subunit (assA) was positive in the enrichment cultures. Moreover, the appearance of a new assA gene sequence identified in day 749 supported the establishment of a functioning microbial species in the enrichment. Our results indicate that n-alkanes are converted to methane slowly by a microbial community enriched from oilfield production water and fumarate addition is most likely the initial activation step of n-alkanes degradation under thermophilic methanogenic conditions.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Anaerobic degradation of petroleum hydrocarbons is generally well documented. Most of the altered petroleum reservoirs have undergone biodegradation to some extents worldwide with anaerobic hydrocarbon biodegradation as a major process involved (Aitken et al. 2004; Duncan et al. 2009; Gieg et al. 2010; Head et al. 2003). Among the electron acceptors available in petroleum reservoirs, methanogenic degradation of crude oil hydrocarbons within these subsurface ecosystems has recently become an important focus for the microbial conversion of residual oil in reservoirs into natural gas (methane) through biological methanogenesis (Gieg et al. 2008; Jones et al. 2008). Indeed, these oil-rich environments represent a significant resource of hydrocarbons for bioconversion by the methanogenic populations. However, only a handful of reports of methanogenic enrichments for degrading crude oil alkanes have been established with production waters from oil reservoirs (Gieg et al. 2010; Wang et al. 2011). Information about microbial communities and the biochemical pathways involved for alkanes-degrading consortium under thermophilic methanogenic conditions is rather scarce (Gieg et al. 2010).
n-Alkanes, a main constituent of petroleum, are biodegraded under methanogenic conditions via syntrophic processes involving both bacteria and archaea. In the biodegradation process, n-alkanes can be initially activated by different strategies, including fumarate addition, carboxylation, or alternative mechanisms (Mbadinga et al. 2011). The initial products after fumarate addition can be used as specific biomarkers since they are only formed under anaerobic condition by alkylsuccinate synthase (Ass) catalyzed reactions. However, detection of alkylsuccinates in n-alkanes-degrading methanogenic enrichments can be a challenge because of their transient nature and very low concentrations. Alternatively, gene encoding the alkylsuccinate synthase α-subunit (assA) can serve as a useful biomarker to track for the presence of functional anaerobic n-alkanes-degrading members (Callaghan et al. 2010).
In the present work, an attempt was made by utilizing oil reservoir production water as inoculums to enrich a high-temperature n-alkanes-degrading methanogenic consortium in order to investigate the microbial community dynamics and the possible biochemical activation mechanisms. 16S rRNA gene-based clone libraries, DGGE and functional genes (assA) were used to reveal the community dynamics over the long-term incubation. Moreover, the recovery of a new assA gene sequence indicated that the n-alkanes-degrading methanogenic enrichment may contain new microbial species.
Materials and methods
Preparation of methanogenic enrichment cultures
Production water from oil producing well at Shengli oilfield of P.R. China was collected and used in the preparation of the thermophilic methanogenic enrichment cultures. Description of the sampling and the water characteristics are reported previously (Wang et al. 2011).
Fifty milliliter of the oil reservoir fluid were transferred aseptically into each 120 ml sterile serum bottle and flushed with O2-free N2 (99.99 %) after passing through heated copper filings. After flushed with N2, the serum bottles were capped with butyl rubber stoppers (Bellco Glass, Inc., Vineland, NJ) and aluminum crimp sealed, and incubated at 55 °C in the dark until substantial methane formation was detected after 100 days by analysis of headspace gas on gas chromatography (GC). These bottles were then flushed with the pure N2 again to remove the methane produced in the headspace; and alkanes-based methanogenic enrichment cultures were then established by transferring 5 ml of the primary incubation culture mentioned above into 120 ml serum bottles, that had been flushed with O2-free N2 (99.99 %), containing each 45 ml mineral salt medium (Wang et al. 2011). After further flushing the serum bottles for 5 min with pure N2, they were sealed with butyl rubber stoppers and aluminum crimps. An equal mixture (v/v) of long chain n-alkanes (C15H32, C16H34, C17H36, C18H38, C19H40 and C20H42, 74.65 μmol total n-alkanes) (Sigma-Aldrich, Milwaukee, WI) was prepared and amended into each enrichment culture before dispensing mineral medium. All incubations including the background controls (without n-alkanes) were prepared in the same way in triplicate. Controls, amended with the n-alkanes mixtures and autoclaved three times successively, were also included. All the microcosms including the controls were incubated at 55 °C in the dark. Changes of microbial community were monitored through sampling of enrichment cultures in the serum bottles. For convenience, B1–B5 and B13 were samples of the alkanes-based methanogenic enrichments collected on day 154, 253, 333, 413, 477 and 749 of incubation, respectively. B11 was the background control (without addition of n-alkanes) sampled on day 749.
Chemical analyses of methanogenic enrichment cultures
Methane and hydrogen gases in the headspace of serum bottles were monitored periodically by injecting 200 μl of the headspace gas into GC equipped with a flame ionization detector (FID), a thermal conductivity detector (TCD), and a packed 1.5 m stainless steel column filled with 5 Å carbon molecular sieves as previously described (Wang et al. 2011).
After taking the incubation cultures for genomic DNA extraction, the aliquot samples were adjusted to pH 12 and then heated for 1 h to 105 °C. n-Alkanes amended into methanogenic enrichment were extracted with n-hexane and analyzed on an Agilent 6890 GC equipped with an HP-5MS capillary column (30 m × 0.25 mm × 0.25 μm) and mass detector (MSD 5975) as described by Wang et al. (2011). Volatile fatty acids (VFA) were detected on GC–MS after butyl esterification of extract with 1:10 H2SO4–butanol (v/v) at 90 °C for 60 min and being extracted with n-dodecane. The injector temperature was 250 °C and the oven temperature was initially held at 60 °C for 1 min, and then increased at 15 °C min−1 to 145 °C. A portion of the above mentioned butyl esterified products were extracted with n-hexane to check for non-volatile fatty acids. GC–MS analysis was accomplished with the following program: the injector temperature was 250 °C and the oven temperature was held at 120 °C for 3 min, then increased at 8 °C min−1 to 260 °C, and held for 10 min.
Simultaneously, 20 ml of enrichment aliquot were adjusted to pH 12, left for 30 min and heated for 1 h to 105 °C before acidification to pH <2 with HCl and being extracted three times with ethyl ether. Pooled organic extracts were dried over anhydrous NaSO4 and concentrated under a stream of N2. The resulting organic extracts were derivatized with N, O-bis (trimethylsilyl) trifluoroacetamide (BSTFA) (Sigma-Aldrich Shanghai Trading Co Ltd, Shanghai, China) at 60 °C for 60 min to introduce trimethylsilyl groups before analysis on GC–MS. The injector temperature was 260 °C and the oven temperature was initially held at 80 °C for 3 min, then increased at 10 °C min−1 to 280 °C, and finally held for 37 min. All analyses were completed in the scan mode. EI was operated at 70 eV and ion source temperature was held at 230 °C.
DNA extraction and PCR amplification
Total genomic DNA was extracted from 10 ml of enrichment culture using AxyPrep™ Bacterial Genomic DNA Miniprep Kit (Axygen Biosciences, Inc., CA, USA) according to the manufacturer’s protocol. Extracted genomic DNAs were immediately frozen and store at −70 °C until further use.
Amplification of 16S rRNA gene for bacteria and archaea
Small-subunit rRNA genes in the pooled DNA samples were amplified using PCR primers 8F (5′-AGAGTTTGATYMTGGCTCAG-3′) and 805R (5′-GACTACCAGGGTATCTAATCC-3′) for bacteria (Savage et al. 2010); primers ARC109F (5′-ACKGCTCAGTAACACGT-3′) and ARC915R (5′-GTGCTCCCCCGCCAATTCCT-3′) specific for archaea (Cheng et al. 2007). Twenty five microliter (25 μl) of PCR reaction mixtures including 13 μl of ddH2O, 9 μl of 2× Taq PCR Master Mix, 1 μl of each primer (12.5 μM) and 1 μl of template DNA, were used. PCR amplifications of the 16S rRNA gene fragments from the genomic DNA were carried out in a Peltier thermal cycler (Bio-Rad, USA). The thermal cycler programs for bacteria and archaea were initial denaturation at 95 °C for 5 min, 33 cycles at 95 °C for 30 s, annealing gradient from 45 to 55 °C (for bacteria) and a single annealing at 60 °C (for archaea) for 30 s, extension at 72 °C for 60 s, and a final elongation step of 72 °C for 10 min.
Amplification of alkylsuccinate synthase alpha-subunit A (assA) gene fragments
Gene encoding the alpha subunit of the alkylsuccinate synthase was amplified with primers 1432F (5′-CCNACCACNAAGCAYGG-3′) and 1933R (5′-TCGTCRTTGCCCCATTTIGGIGC-3′) (Callaghan et al. 2010). Twenty five microliter (25 μl) of PCR reaction mixtures included 7.5 μl of ddH2O, 12.5 μl of 2× Taq PCR Master Mix, 1 μl of each primer (12.5 μM) and 3 μl of template DNA. The thermal cycler program for assA gene included initial denaturation at 95 °C for 5 min; followed by 40 cycles of 95 °C for 36 s, annealing at 60 °C for 60 s, and extension at 72 °C for 2 min; followed by a final extension step at 72 °C for 20 min.
Unless otherwise mentioned, all PCR products obtained above were first visualized by agarose gel (1 %, w/v) electrophoresis followed by ethidium bromide staining to ensure the correct size amplified. Subsequently, PCR products resulting from independent 5 reactions were pooled and visualized by agarose gels (1.8 %, w/v) electrophoresis (70 min at 160 V). The appropriate sizes of these fragments were excised and purified with a DNA purification kit (U-gene, China) prior to cloning.
Amplification of 16S rRNA fragments for bacterial DGGE profile
For bacterial DGGE analysis, genomic DNAs retrieved from methanogenic enrichment cultures were amplified with primer set 341F-GC (Muyzer et al. 1993) and 907R (Muhling et al. 2008). Fifty microliter (50 μl) of PCR reactions included 30 μl of ddH2O, 2× Taq PCR Master Mix 15 μl, 2 μl of each primer (12.5 μM) and 1 μl of template DNA. Thermal cycler conditions were 95 °C for 5 min, 35 cycles at 95 °C for 30 s, annealing at 51 °C for 30 s, extension at 72 °C for 1 min with a final extension at 72 °C for 10 min. PCR products (20 μl) were loaded onto a 6 % (w/v) polyacrylamide gel with a denaturing gradient of 30–60 % (100 % denaturant contained 40 % formamide and 7 M urea). PCR products for DGGE were separated using the DCode™ universal mutation detection system (Bio-Rad, USA). Electrophoresis was performed for 4.5 h at 160 V and 60 °C in TAE running buffer (40 mM Tris–acetate, 20 mM sodium acetate, 1 mM EDTA). DGGE gels were stained with GelRed (Sigma-Aldrich) and photographed under UV transillumination by using a Tanon gel imaging system 2026. Selective bands were excised from the gels, eluted in sterile de-ionized water, and reamplified with the primers set as described above without GC clamp. PCR products were purified with a DNA purification kit (U-gene, China) and sequenced after cloning into Escherichia coli (see below).
Construction of 16S rRNA and assA genes clone libraries
Purified 16S rRNA gene and catabolic gene (assA) fragments were directly cloned into Escherichia coli using a pMD®19-T Simple cloning vector (Takara, Japan) following the instructions of the manufacturer. The obtained white colonies were randomly picked and cultured overnight at 37 °C in 0.8 ml Luria Broth (LB) medium in the presence of ampicillin (100 mg/ml). The size of the gene inserts was checked by PCR using the forward M13F (-47) (5′-CGCCAGGGTTTTCCCAGTCACGAC-3′) and the reverse RV-M (5′-GAGCGGATAACAATTTCACACAGG-3′) plasmid specific primers, followed by agarose gel electrophoresis with subsequent ethidium bromide staining.
Sequencing and phylogenetic analysis
The sequences of inserted PCR products were determined with automated ABI 3730 sequencer (Dye-Terminator Cycle Sequencing; Applied Biosystems) using the forward sequencing primer M13F (−47). After sequencing, reads were first trimmed to remove vector and primer sequences before further analysis. The resulting sequences were checked for orientation by using OrientationChecker tool (Ashelford et al. 2006). Putative chimeric sequences were double checked with Bellerophon 3.0 (Huber et al. 2004) and Pintail (Ashelford et al. 2005). The FastGroupII program (Yu et al. 2006) was used to group non-chimeric sequences into operational taxonomic units (OTUs) at a cut-off of 97 % with gaps. A single sequence was chosen as representative of each OTU for phylogenetic tree construction. The classifier tool of the Ribosomal Database Project II (Wang et al. 2007) was used to classify each 16S rRNA gene sequence. The nearest relatives of each OTU were identified using the NCBI BLAST network service at http://www.ncbi.nlm.nih.gov/blast/ (Altschul et al. 1990; McGinnis and Madden 2004). The coverage of each 16S rRNA gene clone libraries was calculated by the equation C = [1 − (n 1/N)] × 100 (Good 1953). Phylogenetic trees were constructed based on the Kimura two-parameter model (Kimura 1980) and neighbor-joining algorithm (Saitou and Nei 1987) for nucleotide sequences and the Poisson correction method for amino acid sequences using the MEGA5 software (Tamura et al. 2011). Bootstrap analysis with 1000 replicates was applied to assign confidence levels to the nodes in the trees. Partial 16S rRNA gene sequences for bacterial and archaeal as well as assA genes fragments obtained in this study were deposited in the GenBank database under accession numbers JN836375–JN836429 and JN852952–JN852958.
Results
Methanogenic activities in alkanes-based enrichment cultures
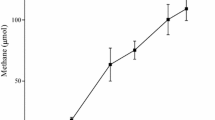
Thermophilic methanogenic activity in enrichment cultures containing production water of a thermophilic oil reservoir was observed with significant amounts of methane produced in headspace during the 749 days of incubation after transferring into fresh culture media supplemented with long chain n-alkanes (Fig. 1). Sterile controls did not show any evidence of gas formation throughout the incubation. GC–MS analysis of enrichment cultures medium showed that approximately 83 % n-alkanes were transformed during the 749 days of incubation compared with the sterile controls. Based on the amount of n-alkanes consumption, about 828.91 μmol of methane should have been theoretically produced as estimated from stoichiometric equations (Symons and Buswell 1933; Zengler et al. 1999). However, approximately 141.47 μmol of methane; corresponding to 17 % of the theoretical value, were obtained in the bottles headspace.

Methane production in enrichment cultures amended with a mixture of n-alkanes (closed squares) and Shengli oilfield production water incubated at 55 °C, compared with inoculated controls without any n-alkanes (open squares). Hydrogen gas production in the enrichement with n-alkanes (closed circles) and without any n-alkanes (open circles). Errors show the standard deviation of triplicate samples
Metabolites formed during methanogenic n-alkanes degradation
A series of higher alkanoic acids that included tetracosanoic acid (C24), docosanoic acid (C22), eicosanoic acid (C20), octadecanoic acid (C18), palmitic acid (C16), pentadecanoic acid (C15), tetradecanoic acid (C14), dodecanoic acid (C12), undecylic acid (C11), capric acid (C10), nonanoic acid (C9) and octanoic acid (C8) were detected with concentrations ranging from 0.69 to 82.67 μM in the enrichment cultures of the alkanes-amended. These compounds might represent downstream metabolites of the initial activation and fermentation of n-alkanes (Mbadinga et al. 2011; Zhou et al. 2011). Volatile fatty acids (C1–C4) with concentrations ranging from 1.38 to 72.32 μM were also detected in the enrichment cultures. In addition, a dicarboxylic acid referring to as fumarate was detected. The detection of these metabolic intermediates provided strong chemical evidences on the incomplete conversion of substrate n-alkanes to methane. On the other hand, it indicates that n-alkanes were activated and transformed by the enriched methanogenic microbial community.
Microbial community in thermophilic methanogenic alkanes-based enrichment cultures
Genomic DNAs extracted from thermophilic methanogenic enrichment cultures were subjected to molecular analyses in order to determine the microbial community compositions in these enrichment cultures.
Archaeal composition
A total of 90 clones were sequenced from sample B1 (day 154 after incubation). These clones were clustered into 16 OTUs at 97 % similarity cut-off and rarefaction analysis indicated that the estimated coverage of the clone library was 91.1 % (Fig. 2). Phylogenetic analysis revealed that approximately 45.6 % of the clones in this library were affiliated with the order Methanosarcinales, which is known as a group harboring both acetotrophic and methylotrophic methanogens (Kendall and Boone 2006). In the present case, methylotrophic methanogens represented by OTUs B1-A-103, B1-A-41 and B1-A-64 and closely related (95–99 % similarities) to the genus Methanomethylovorans prevailed. Phylotypes clustering with the orders Methanobacteriales and Methanomicrobiales were detected in relatively low abundance. Members of these orders can produce methane by CO2-reduction or alternatively from formate (Bonin and Boone 2006; Garcia et al. 2006). The remaining phylotypes, representing 48.9 % of the total archaeal clones in library B1, were assigned to unclassified groups of Euryarchaeota and Crenarchaeota. Most clones in these groups were related to environmental 16S rRNA gene sequences from oil reservoirs and hydrocarbons contaminated ecosystems (Fig. 2).

Phylogenetic tree of archaeal clones obtained from the alkanes-based methanogenic enrichment cultures taken at different incubation times with closely related sequences. Evolutionary analyses were conducted with MEGA5 software. The topology shown was obtained with the neighbor-joining method. Bootstrap values (n = 1,000 replicates) of ≥65 % are reported as percentages. B1 and B13 included 90 and 89 sequences, respectively. Sequences from organisms identified in hydrocarbons-rich environments or those from hydrocarbons degrading enrichment cultures are shown in bold
After another 600 days of incubation (Sample B13, day 749), the archaeal community experienced some changes. A total of 89 clones were sequenced and clustered into 13 OTUs at 97 % similarity cut-off and rarefaction analysis indicated that the estimated coverage of the clone library was 95.5 % (Fig. 2). Unexpectedly, phylogenetic analysis revealed the dominance of Crenarchaeota-like (52.8 % of the total archaeal clones, 4 OTUs) members mostly affiliated with environmental sequences retrieved from subsurface ecosystems (Duncan et al. 2009; Gihring et al. 2006) or with sequences detected in hydrocarbon-associated methanogenic consortia (Penner and Foght 2010; Siegert et al. 2011). The phylum Euryarchaeota was essentially represented by methanogens clustering with the orders Methanosarcinales (2 phylotypes; 9.0 %), Methanobacteriales (4 phylotypes, 31.5 %) and Methanomicrobiales (2 phylotypes; 5.6 %) in addition to the presence of a single phylotype belonging to the order Halobacteriales. Among the Euryarchaeota, archaeal sequences affiliated with the genus Methanothermobacter within the order Methanobacteriales prevailed.
Bacterial community and composition
Bacterial community changes were first elucidated by means of PCR-DGGE approach (Fig. 3). Selective DNA bands from the DGGE profile were excised, cloned and sequenced. Results showed the presence of Firmicutes-like sequences (bands 1 and 3) closely affiliated to Moorella and Gelria spp. Also sequenced was band 2 affiliated with members of the Candidate division OP8. DGGE band 1 (Moorella sp.) was detected in all profiles whereas band 3 (Gelria sp.) was unique to the alkanes-based methanogenic enrichment cultures.

PCR-based DGGE fingerprints of the bacteria from methanogenic enrichment sampling points at different incubation times (B1–B5, B13 and B11, described in “Materials and methods” for more detail). The denaturing gradient range was from 30 to 60 %. The randomly selected sequences of bands indicated by numbers were aligned and showed the presence of Moorella sp., a member of the candidate division OP8 as well as Gelria sp. The results corresponded to the bacterial phylogenetic tree
Following changes observed from the DGGE profiling (Fig. 3), bacterial community compositions were assessed by means of 16S rRNA gene clone libraries focused on samples B1 and B13 collected at days 154 and 749, respectively. Within B1, 19 clones were sequenced and clustered into 4 bacterial OTUs and the estimated coverage of 89.5 % indicated that the bacterial composition needed more sampling for higher coverage (Fig. 4). Phylogenetic analysis revealed that, of the 19 clones from the B1 library, OTU B1-B-65 consisting of 15 clones (78.9 % of total bacterial clones within B1) was a close relative to Moorella glycerini (95 % identity) within the family Thermoanaerobacteraceae. Moorella spp. are thermophilic obligates anaerobes generally known for syntrophic metabolism (Pierce et al. 2008; Slobodkin et al. 1997) with a growth temperatures ranging from 40 to 70 °C and an optimum between 55 and 60 °C (Balk et al. 2008; Slobodkin et al. 1997). Among other bacterial members in library B1, 16S rRNA gene sequences clustering with members of Thermodesulfobiaceae (OTU B1-B-62, 2 clones) and Leptospiraceae and Bacteroidetes (OTUs B1-B-33 and B1-B-73, 2 clones in total) were also detected.

Phylogenetic tree of bacterial clones obtained from the alkanes-amended methanogenic enrichment cultures at different incubation times with closely related sequences. Evolutionary analyses were conducted with MEGA5 software. The topology shown was obtained with the neighbor-joining method. Bootstrap values (n = 1,000 replicates) of ≥65 % are reported as percentages. B1 represented 19 sequences, while B13 represented 170 sequences. Sequences from organisms identified in hydrocarbons-rich environments or those from hydrocarbons degrading enrichment cultures are shown in bold
In contrast to B1, the bacterial community composition in clone library B13 (n = 170 clones) was slightly more diverse with 8 OTUs detected and the estimated coverage of the clone library of 97.6 % indicated adequate sampling of bacterial members in sample B13 (Fig. 4). The B13 library was dominated by Firmicutes-like members; among which OTUs B13-B-83, B13-B-68 and B13-B-27 representing 90 clones (52.9 % of total bacterial clones within B13) showing high identity with the thermophilic, obligately syntrophic anaerobe Gelria glutamica. Optimal temperature of growth for this cultured representative of the genus Gelria is between 50 and 55 °C (Plugge et al. 2002). The second dominant bacterial members were represented by phylotypes OTUs B13-B-72 and B13-B-103 (44.1 % of total bacterial clones within B13, 75 clones) sharing close similarities with members of the genus Moorella. Other bacterial phylotypes in the B13 clone library comprised members of the Thermotogae, Chloroflexi, as well as a member of the Candidate division OP1, all together representing 5 clones (3.0 % of total bacterial clones within library B13).
Alkylsuccinate synthase alpha-subunit A (assA)
Genomic DNAs extracted from enrichment cultures taken at different incubation times were screened for the presence of genes encoding the alkylsuccinate synthase alpha-subunit A (assA). A total of 80 clones retrieved from library B13 were sequenced and clustered into 2 OTUs (Fig. 5). The absence of stop codons suggests that the deduced amino acid products are partial sequence of the functional Ass proteins. OTU B13-assA-1 representing 78 clones (97.5 % of total assA genes in library B13) was 77 % similar at the protein level to deduced reference assA genes from the Syntrophobacteraceae, alkanes-degrader Desulfoglaeba alkanexedens ALDC (Davidova et al. 2006) and methanogenic paraffin degrading enrichment culture (Callaghan et al. 2010). Noteworthy, sequences highly similar to OTU B13-assA-1 were also identified in samples B1 and B11 (Fig. 5). In addition, two clones (2.5 % of total assA genes in library B13) represented by OTU B13-assA-2 and exclusively detected in the n-alkanes-amended methanogenic cultures are close relatives (83 % similarity at the protein level) to the deduced assA1 gene from the Desulfobacteraceae and syntrophic alkanes degrader Desulfatibacillum alkenivorans AK-01 (Callaghan et al. 2008, 2012).

Phylogenetic tree of deduced assA gene sequences from the methanogenic alkanes-degrading enrichment at different incubation times with closely related sequences. Evolutionary analyses were conducted with MEGA5 software. The topology shown was obtained with the neighbor-joining method. Bootstrap values (n = 1,000 replicates) of 65 % are reported as percentages. B1 represented 57 sequences; B13 represented 80 sequences; B11 represented 1 sequence
Discussion
Partial conversion of n-alkanes to methane
Anaerobic degradation of n-alkanes has been reported in enriched cultures from near surface sediments (Zengler et al. 1999). Little has been reported with samples originating from petroleum reservoirs, especially those collected from high-temperature subsurface oil-rich ecosystems (Gieg et al. 2010). It is generally accepted that methanogenic degradation of alkanes is a slow process resulting in slow growth and low conversion rates due to substrate activation and syntrophic metabolism. Though higher amounts of methane were detected in the n-alkanes-amended methanogenic cultures in comparison to the background controls, stoichiometric estimation indicated only 17 % of the theoretically predicted methane production on day 749 of incubation. This might be caused by potential losses of methane during repeated samplings and the utilization of organic carbon for cellular synthesis. More importantly, incomplete conversion of n-alkanes to methane should be the most important factor as detection of potential downstream metabolites of the anaerobic degradation of n-alkanes in the enrichment cultures was evident. The reasons for the accumulation of these metabolites to detectable level are not well understood. Nutritional limitation and/or inhibitors buildup cannot be totally ruled out. Therefore, for accurate stoichiometric estimation it is necessary to take into account the mass of metabolites accumulated in the culture medium. In that case, the ratio between the produced and theoretically predicted methane would have been much higher than 17 % calculated assuming that the n-alkanes were totally converted to methane.
Community dynamics and methanogenesis
Methanogenic degradation of n-alkanes has been reported before, but the present study was designed to assess dynamic changes of microbial communities during long-term incubation. Considering bacterial clone library of the alkanes-based methanogenic enrichment, the earlier phylogenetic profile (day 154) was dominated by Moorella spp., similar to that of the background control OTU B11-B-11 (Fig. S1). In contrast, the bacterial population profile retrieved from the n-alkanes amended methanogenic culture at day 749 was quite different. The only phylotype present in substantial proportions in both the earlier and the later settings was Moorella sp. Other sequences retrieved from day 749 were very different and form clusters with Gelria, Thermotogaceae, Chloroflexi, and OP1 (Fig. 6). To our knowledge, only a few reports have shown the occurrence of Moorella spp. in production water from oil reservoirs (Duncan et al. 2009; Mayumi et al. 2011) whereas Gelria spp. have never been reported. Furthermore, none of the bacterial phylotypes identified are known to degrade n-alkanes anaerobically. However, there are reports showing that methanogenic biodegradation of alkanes or crude oil alkanes involve acetogenic bacteria, syntrophs and methanogens (Jones et al. 2008; Zengler et al. 1999). These assumptions are consistent with the detection of Moorella spp. as well as the later emergence of Gelria spp. in the alkanes-amended methanogenic enrichment culture. On the other hand, with the exception of the phylotype represented by B13-A-4, most of the Crenarchaeota identified in B13 were not detected in B1 (Fig. 2). This observation indicates the enrichment of new Crenarchaeota phylotypes following the progressive anaerobic degradation of n-alkanes under thermophilic methanogenic conditions. For instance, Crenarchaeota are not known to produce methane. Therefore, they could have been implicated in the syntrophic conversion of organic carbon to methane. This assumption is also supported by the low frequency of Crenarchaeota-like sequences in the control incubation OTU B11-A-106 (only one sequence was detected, Fig. S2). Using the classifier tool of the RDP Release 10 (Cole et al. 2009), most of the Crenarchaeota-like sequences identified in our thermophilic alkanes-based methanogenic enrichment cultures appear to be related to members of the class Thermoprotei; but represented by low similarities to known cultured species.

Dynamic changes of bacterial community composition in the alkanes-amended methanogenic enrichment cultures on days 154 and 749
Dynamic changes observed with assA genes suggest that the thermophilic alkanes-amended methanogenic enrichment cultures harbor microorganisms likely involved in the anaerobic degradation of n-alkanes via fumarate addition. It is important to mention that, in the alkanes-amended methanogenic enrichment culture assA gene with close affiliation to assA1 gene from Desulfatibacillum alkenivorans AK-01 was detected in this study. However, 16S rRNA sequences affiliated to members of the Proteobacteria were not found in the methanogenic enrichment cultures. This has implication for the role played by microorganisms other than Proteobacteria in the thermophilic anaerobic activation of n-alkanes via fumarate addition.
Anaerobic conversion of n-alkanes to methane requires the close cooperation between metabolically different types of microorganisms. The overall biodegradation process is thermodynamically feasible only when methanogenic precursors, namely formate, acetate and hydrogen gas, are completely removed or maintain at very low concentration by methanogens or sulfidogens. There are several routes whereby n-alkanes are converted to methane; with the predominant occurrence of the MADCOR (methanogenic alkanes degradation dominated by CO2 reduction) process in petroleum reservoirs (Dolfing et al. 2008; Jones et al. 2008). The methanogenic population in our thermophilic alkanes-based enrichment cultures exhibited considerable changes from methylotrophic/acetoclastic to CO2-reducing phylotypes (Fig. 7). Our findings are consistent with several reports indicating that alkanes are predominantly degraded to acetate and hydrogen with subsequent syntrophic acetate oxidation to hydrogen and CO2 coupled with hydrogenotrophic methanogenesis (Gieg et al. 2010; Siddique et al. 2011).

Dynamic changes of methanogens in the alkanes-amended methanogenic enrichment cultures on days 154 and 749
Anaerobic activation of n-alkanes and putative biochemical degradation pathways
There are several possible biochemical activation strategies of n-alkanes under anaerobic conditions. However, addition of n-alkanes to the double bond of fumarate appears to be the most widely spread activation strategy. As depicted in Fig. 8, though alkylsuccinates were not detectable in the enrichment cultures (probably due to the small volume of the cultures available or non accumulation of alkylsuccinates in the methanogenic cultures), the detection of genes encoding the alkylsuccinate synthase alpha-subunit A (assA) in the alkanes-amended methanogenic enrichment cultures indicates that fumarate addition was likely involved in the initial activation of n-alkanes. Further degradation of alkylsuccinates will result in the formation of branched and linear fatty acids (Callaghan et al. 2012). However, alkylsuccinates and other branched fatty acids were not detectable in the alkanes-based methanogenic enrichment cultures of this study, while a series of linear fatty acids representing possible downstream metabolites of alkylsuccinates were detected. Therefore, the biochemical degradation pathways that could have been involved in the anaerobic conversion of n-alkanes to methane are proposed in the present work (Fig. 8). Alkanes were most likely activated by addition to fumarate and subsequently biodegraded via fatty acids by members of the Firmicutes and Crenarchaeotes into acetate, formate, H2 and CO2 which in turn are consumed by acetoclastic (Methanosarcinales) and hydrogenotrophic (Methanomicrobiales and Methanobacteriales) methanogens into methane.

Proposed biochemical degradation pathways in the anaerobic conversion of n-alkanes to methane. (1) Fumarate addition (Callaghan et al. 2012); (2) Dehydrogenation and hydroxylation (Mbadinga et al. 2011); (3) Alternative mechanisms. The red circles indicate metabolites detected in the methanogenic alkane-biodegradation, while the grey boxes show the intermediates were not detected (Color figure online)
Alternatively, part of the acetate formed during the anaerobic degradation of n-alkanes should be syntrophically oxidized to H2 and CO2 followed by methanogenesis from CO2-reduction and acetoclastic methanogenesis. This assumption is supported by the fact that CO2-reducing methanogens (Methanomicrobiales and Methanobacteriales) outnumbered obligate acetoclastic methanogens and represented >80 % of methanogenic archaeal sequences detected in the thermophilic alkanes-amended methanogenic cultures on day 749 (Fig. 7).
Conclusion
In the present work, a thermophilic methanogenic alkanes-degrading community enriched from high-temperature oil reservoirs production fluid contained the assA genes indicating that fumarate addition was likely involved in the initial activation of n-alkanes. The presence of abundant members of the Firmicutes (Moorella and Gelria) implicated their roles contributing to anaerobic biodegradation of n-alkanes. Also, organisms affiliated to the Crenarchaeotes would have played an important role in the methanogenic conversion of n-alkanes to methane. At the same time, the accumulation of intermediate metabolites is indicative of a partial conversion of n-alkanes to methane in the current system. The current study offers further information on our understanding of the methanogenic biodegradation biochemical processes and functional microorganisms involved in the anaerobic degradation of alkanes in subsurface petroleum reservoirs.
References
Aitken CM, Jones DM, Larter SR (2004) Anaerobic hydrocarbon biodegradation in deep subsurface oil reservoirs. Nature 431:291–294
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Ashelford KE, Chuzhanova NA, Fry JC, Jones AJ, Weightman AJ (2005) At least 1 in 20 16S rRNA sequence records currently held in public repositories is estimated to contain substantial anomalies. Appl Environ Microbiol 71:7724–7736
Ashelford KE, Chuzhanova NA, Fry JC, Jones AJ, Weightman AJ (2006) New screening software shows that most recent large 16S rRNA gene clone libraries contain chimeras. Appl Environ Microbiol 72:5734–5741
Balk M, van Gelder T, Weelink SA, Stams AJM (2008) (Per)chlorate reduction by the thermophilic bacterium Moorella perchloratireducens sp. nov., isolated from underground gas storage. Appl Environ Microbiol 74:403–409
Bonin AS, Boone DR (2006) The order methanobacteriales. In: Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E (eds) The prokaryotes, 3rd edn. Springer, New York, pp 231–243
Callaghan AV, Wawrik B, Ní Chadhain SM, Young LY, Zylstra GJ (2008) Anaerobic alkane-degrading strain AK-01 contains two alkylsuccinate synthase genes. Biochem Biophys Res Commun 366:142–148
Callaghan AV, Davidova IA, Savage-Ashlock K, Parisi VA, Gieg LM, Suflita JM, Kukor JJ, Wawrik B (2010) Diversity of benyzl- and alkylsuccinate synthase genes in hydrocarbon-impacted environments and enrichment cultures. Environ Sci Technol 44:7287–7294
Callaghan AV, Morris BEL, Pereira IAC, McInerney MJ, Austin RN, Groves JT, Kukor JJ, Suflita JM, Young LY, Zylstra GJ, Wawrik B (2012) The genome sequence of Desulfatibacillum alkenivorans AK-01: a blueprint for anaerobic alkane oxidation. Environ Microbiol 14:101–113
Cheng L, Qiu T-L, Yin X-B, Wu X-L, Hu G-Q, Deng Y, Zhang H (2007) Methermicoccus shengliensis gen. nov., sp. nov., a thermophilic, methylotrophic methanogen isolated from oil-production water, and proposal of Methermicoccaceae fam. nov. Int J Syst Evol Microbiol 57:2964–2969
Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM (2009) The ribosomal database project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res 37:D141–145
Davidova IA, Duncan KE, Choi OK, Suflita JM (2006) Desulfoglaeba alkanexedens gen. nov., sp. nov., an n-alkane-degrading, sulfate-reducing bacterium. Int J Syst Evol Microbiol 56:2737–2742
Dolfing J, Larter SR, Head IM (2008) Thermodynamic constraints on methanogenic crude oil biodegradation. ISME J 2:442–452
Duncan KE, Gieg LM, Parisi VA, Tanner RS, Tringe SG, Bristow J, Suflita JM (2009) Biocorrosive thermophilic microbial communities in Alaskan North Slope oil facilities. Environ Sci Technol 43:7977–7984
Garcia J-L, Ollivier B, Whitman WB (2006) The order Methanomicrobiales. In: Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E (eds) The prokaryotes, 3rd edn. Springer, New York, pp 208–230
Gieg LM, Duncan KE, Suflita JM (2008) Bioenergy production via microbial conversion of residual oil to natural gas. Appl Environ Microbiol 74:3022–3029
Gieg LM, Davidova IA, Duncan KE, Suflita JM (2010) Methanogenesis, sulfate reduction and crude oil biodegradation in hot Alaskan oilfields. Environ Microbiol 12:3074–3086
Gihring TM, Moser DP, Lin L-H, Davidson M, Onstott TC, Morgan L, Milleson M, Kieft TL, Trimarco E, Balkwill DL, Dollhopf ME (2006) The distribution of microbial taxa in the subsurface water of the Kalahari Shield, South Africa. Geomicrobiol J 23:415–430
Good IJ (1953) The population frequencies of species and the estimation of population parameters. Biometrika 40:237–264
Head IM, Jones DM, Larter SR (2003) Biological activity in the deep subsurface and the origin of heavy oil. Nature 426:344–352
Huber T, Faulkner G, Hugenholtz P (2004) Bellerophon: a program to detect chimeric sequences in multiple sequence alignments. Bioinformatics 20:2317–2319
Jones DM, Head IM, Gray ND, Adams JJ, Rowan AK, Aitken CM, Bennett B, Huang H, Brown A, Bowler BFJ, Oldenburg T, Erdmann M, Larter SR (2008) Crude-oil biodegradation via methanogenesis in subsurface petroleum reservoirs. Nature 451:176–180
Kendall MM, Boone DR (2006) The order Methanosarcinales. In: Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E (eds) The prokaryotes, 3rd edn. Springer, New York, pp 244–256
Kimura M (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120
Mayumi D, Mochimaru H, Yoshioka H, Sakata S, Maeda H, Miyagawa Y, Ikarashi M, Takeuchi M, Kamagata Y (2011) Evidence for syntrophic acetate oxidation coupled to hydrogenotrophic methanogenesis in the high-temperature petroleum reservoir of Yabase oil field (Japan). Environ Microbiol 13:1995–2006
Mbadinga SM, Wang L-Y, Zhou L, Liu J-F, Gu J-D, Mu B-Z (2011) Microbial communities involved in anaerobic degradation of alkanes. Int Biodeter Biodegrad 65:1–13
McGinnis S, Madden TL (2004) BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res 32:W20–W25
Muhling M, Woolven-Allen J, Murrell JC, Joint I (2008) Improved group-specific PCR primers for denaturing gradient gel electrophoresis analysis of the genetic diversity of complex microbial communities. ISME J 2:379–392
Muyzer G, de Waal EC, Uitterlinden AG (1993) Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol 59:695–700
Penner TJ, Foght JM (2010) Mature fine tailings from oil sands processing harbour diverse methanogenic communities. Can J Microbiol 56:459–470
Pierce E, Xie G, Barabote RD, Saunders E, Han CS, Detter JC, Richardson P, Brettin TS, Das A, Ljungdahl LG, Ragsdale SW (2008) The complete genome sequence of Moorella thermoacetica (f. Clostridium thermoaceticum). Environ Microbiol 10:2550–2573
Plugge CM, Balk M, Zoetendal EG, Stams AJ (2002) Gelria glutamica gen. nov., sp. nov., a thermophilic, obligately syntrophic, glutamate-degrading anaerobe. Int J Syst Evol Microbiol 52:401–407
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Savage KN, Krumholz LR, Gieg LM, Parisi VA, Suflita JM, Allen J, Philp RP, Elshahed MS (2010) Biodegradation of low-molecular-weight alkanes under mesophilic, sulfate-reducing conditions: metabolic intermediates and community patterns. FEMS Microbiol Ecol 72:485–495
Siddique T, Penner T, Semple K, Foght JM (2011) Anaerobic biodegradation of longer-chain n-alkanes coupled to methane production in oil sands tailings. Environ Sci Technol 45:5892–5899
Siegert M, Cichocka D, Herrmann S, Gründger F, Feisthauer S, Richnow H–H, Springael D, Krüger M (2011) Accelerated methanogenesis from aliphatic and aromatic hydrocarbons under iron- and sulfate-reducing conditions. FEMS Microbiol Lett 315:6–16
Slobodkin A, Reysenbach A-L, Mayer F, Wiegel J (1997) Isolation and characterization of the homoacetogenic thermophilic bacterium Moorella glycerini sp. nov. Int J Syst Bacteriol 47:969–974
Symons GE, Buswell AM (1933) The methane fermentation of carbohydrates. J Am Chem Soc 55:2028–2036
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267
Wang L-Y, Gao C-X, Mbadinga SM, Zhou L, Liu J-F, Gu J-D, Mu B-Z (2011) Characterization of an alkane-degrading methanogenic enrichment culture from production water of an oil reservoir after 274 days of incubation. Int Biodeter Biodegrad 65:444–450
Yu Y, Breitbart M, McNairnie P, Rohwer F (2006) FastGroupII: a web-based bioinformatics platform for analyses of large 16S rDNA libraries. BMC Bioinformatics 7:57
Zengler K, Richnow HH, Rosselló-Mora R, Michaelis W, Widdel F (1999) Methane formation from long-chain alkanes by anaerobic microorganisms. Nature 401:266–269
Zhou L, Mbadinga SM, Wang L, Liu J, Yang S, Mu B (2011) Recent progress in metabolites formed during anaerobic biodegradation of petroleum hydrocarbons. Chin J Appl Environ Biol 17:607–613
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Nos. 41073055, 51174092) and NSFC/RGC Joint Research Fund (No. 41161160560).
Conflict of interest
The authors did not report any conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Zhou, L., Li, KP., Mbadinga, S.M. et al. Analyses of n-alkanes degrading community dynamics of a high-temperature methanogenic consortium enriched from production water of a petroleum reservoir by a combination of molecular techniques. Ecotoxicology 21, 1680–1691 (2012). https://doi.org/10.1007/s10646-012-0949-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10646-012-0949-5