
Abstract
A blue protein present in the mucus coating blue forms of walleye, Sander vitreus, was purified to homogeneity by a combination of ion exchange and hydrophobic interaction chromatography. The purified protein has a molecular mass of 87,850 and is a homotetramer with a subunit molecular mass of 21,836. Solutions of the protein are deep blue in color and show absorbance maxima at 383 and 633 nm, respectively. Acetone treatment of the protein releases a blue chromophore with the spectral characteristics of biliverdin IXα. The results suggest that the blue protein is a new biliprotein that we have termed Sandercyanin. The function of Sandercyanin is unknown, but limited amino acid sequence analysis suggests it is a lipocalin that may be involved in the transport of heme degradation products.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Production of color in fish generally is limited to the dermal and epidermal regions of skin and is formed by specialized pigment-producing cells (Fujii 1993). In the case of walleye, Sander vitreus, yellow and blue phenotypes have been described, although the nature of the pigments involved have not been reported. Recently, Paradis and Magnan (2005) reported the presence of blue walleye morphotypes in five lakes in central Quebec, Canada. The fish were covered with a blue mucus, a phenomenon previously seen in blue walleye forms present in The Great Lakes and Lake Nipissing, Canada (Regier et al. 1969; Scott and Crossman 1973). The cause and evolutionary significance of the blue color has not been determined. In this communication we describe and name a novel blue protein, Sandercyanin, present in the mucus of blue walleye populations in northwestern Ontario.
Materials and methods
Blue and yellow walleye forms were captured by hook and line from the Papaonga River system in northwestern Ontario. Mucus was carefully removed from the dorsal side of the fish with a knife and mixed with approximately three volumes of lake water. Samples were stored at 4°C until analyzed.
Protein purification
All chromatographic procedures were performed in a cold room at 4°C using an automated fast protein chromatography system (Bio-Rad Laboratories, Hercules, CA). Chromatography columns and column supports were from Amersham Biosciences, Piscataway, NJ, except for ceramic hydroxyapatite, which was obtained from Bio-Rad Laboratories.
The diluted mucus solution (110 ml) from blue walleye forms was centrifuged at 15,800g for 15 min and the residue discarded. Ammonium sulfate (32 g) was slowly added to the supernatant until the mixture became turbid. After 1 h at 4°C a white precipitate formed which was removed by centrifugation at 13,000g for 10 min. Ammonium sulfate (18 g) was added to the resulting clear blue supernatant solution followed by centrifugation at 13,000g for 15 min. The blue precipitate was dissolved in 35 ml of Tris buffer (50 mM Tris–HCl buffer, pH 7.0). Ammonium sulfate was removed by ultrafiltration. The concentrated solution (168 mg protein) was applied to a Q-Sepharose column (2.6 × 40 cm2) that had previously been equilibrated with Tris buffer. The column was washed with 200 ml of the same buffer at a flow rate of 1.0 ml per min. Bound protein was eluted with a 600 ml linear gradient (0.0–0.5 M) of KCl in Tris buffer. Blue-colored fractions, eluting at ∼0.3 M KCl, were pooled and concentrated to 4.8 ml (10.1 mg of protein) by ultrafiltration. Ammonium sulfate (4.0 M) was added to the concentrated solution to give a final concentration of 1.0 M. The solution was clarified by centrifugation at 13,000g for 10 min and applied to the top of a Phenyl Sepharose High-Performance Column (5.0 × 1.0 ml2) that had previously been equilibrated with Tris buffer containing 1.0 M (NH4)2SO4. The column was washed with the same buffer at a flow rate of 1.0 ml per min until protein was no longer detected in the eluate. Bound protein was eluted with a linear gradient of (NH4)2SO4 (1.0–0.0 M) in the same buffer. Blue-colored fractions were pooled and concentrated by ultrafiltration. Ammonium sulfate was removed by buffer exchange with 5.0 mM KH2PO4, pH 7.0. This preparation (5.0 mg of protein) was applied to a ceramic hydroxyapatite XK16/70 column (80 × 1.0 ml2) that had been equilibrated with 5.0 mM KH2PO4 buffer, pH 7.0. Unbound protein was eluted with 100 ml of the same buffer at a flow rate of 1.0 ml per min before initiating a linear gradient (5–200 mM) of KH2PO4 buffer, pH 7.0. Dark blue fractions (1.0 ml) eluting from the column were pooled, concentrated by ultrafiltration and exchanged into 10 mM KH2PO4 buffer, pH 7.0, before adding in 30 μl aliquots to liquid nitrogen. The resulting pellets were stored at −70°C.
Protein determination
Protein concentrations were determined by the method of Bradford (Bradford 1976) using bovine serum albumin as the standard.
Electrophoresis
Protein purity and subunit molecular weights were determined by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) on 12.5% gels (Laemmli 1970). Samples were boiled for 6 min prior to loading onto the gels. Protein standards used for the estimation of subunit molecular masses were Low-Range Molecular Weight SDS-PAGE Standards (Bio-Rad Laboratories). Protein bands were stained with Coomassie Brilliant Blue R250.
Molecular mass
The molecular mass and subunit molecular mass of the blue protein was determined by matrix-assisted laser desorption ionization-time of flight/mass spectrometry (MALDI-TOF/MS) at The University of Iowa Molecular Analysis Facility, using α-cyano-4-hydroxycinnamic acid (CHCA) in 50% acetonitrile: 50% 0.1% trifluoroacetic acid as matrix.
Cyanogen bromide cleavage
A small crystal of cyanogen bromide was added to 50 μl of formic acid (70%) containing purified blue protein (15 μg). The reaction mixture was incubated in the dark at 25°C for 24 h and then taken to dryness.
Proteolytic digestion
In separate experiments, purified blue protein was digested with endoproteinases Glu-C, Arg-C, Lys-C, and Asp-C, at ratios of endoproteinase to blue protein recommended by the supplier (Sigma Chemical Co., St. Louis, MO). Reaction mixtures were incubated at 37°C for 2–18 h and then taken to dryness.
Amino acid sequence determination
Peptides produced by cyanogen bromide cleavage and proteolytic digestion were separated by SDS-PAGE, electroblotted onto polyvinylidene membranes and their respective amino acid sequences determined by Edman degradation on an automated amino acid sequencer (Applied Biosystems, Foster City, CA) at The University of Iowa Molecular Analysis Facility.
Transition metals
Transition metals were investigated by inductively coupled plasma mass spectrometry (ICP-MS) on a VG PlasmaQuad II ICP-MS machine at the University of Iowa Hygienic Laboratory, Des Moines, IA.
Results
Distribution of blue mucus in walleye
Blue walleye forms taken from the upper reaches of the Papaonga River system near Ear Falls, in northwestern Ontario, were coated with a blue mucus (Fig. 1A). These fish were albino for yellow skin pigment and, after removal of the mucus layer, appeared steel gray in color. The lack of yellow pigment in the membranes of the dorsal and caudal fins made the structures translucent and allowed clear observation of the blue mucus on the fins giving the fish a blue appearance. In each specimen, blue mucus was located only on the dorsal side of the body and was particularly abundant in the rays and interspinous membranes of the two dorsal fins and the caudal fin. Subsequent research in the same location showed blue mucus was also present on yellow walleye forms and apparent hybrids that were mottled for yellow color. Nuclear and mitochondrial DNA analyses showed that the blue and yellow forms were not genetically distinguishable. Moreover, the morphology of the blue form did not match any of the morphological characters that distinguish the blue ‘pike’, Sander vitreus glaucus, and instead matched the morphology of the walleye, Sander vitreus vitreus (C. Stepien, personal communication).

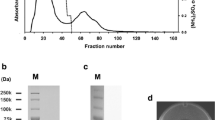
(A) Mucus from blue walleye forms prior to purification. (B) Solution of purified blue protein obtained after hydroxyapatite chromatography (Table 1)
As the study progressed we established a web siteFootnote 1 where fishermen could report sightings of blue walleye forms. To date, blue forms have been identified throughout northwestern Ontario, central Quebec and northern Saskatchewan and have also been identified in several lakes in Minnesota, Wisconsin, and Michigan. In addition, we have taken blue forms of yellow perch, Perca flavescens, from the Papaonga River in Ontario and Winnibigoshish Lake in northern Minnesota.
Purification and characterization of the blue pigment
A blue protein was subsequently purified from the mucus of blue walleye forms collected in the summer of 2003 from the Papaonga River system. The procedure involved a combination of ion exchange and hydrophobic interaction chromatography, and yielded 2.8 mg of a blue protein (Fig. 1B) with a recovery of 19.5% (Table 1). The purified protein was very stable and could be stored at 4°C for up to 8 weeks with no apparent degradation.
Properties of the blue protein
When analyzed by SDS-PAGE, the protein migrated as a single band with a molecular mass of ∼21,500 (Fig. 2A, lanes 5, 6). A single band was observed on non-denaturing gels (Fig. 2B, lanes 2, 3). The molecular masses of the native and SDS-treated protein, determined by MALDI-TOF/MS, were 87,850 and 21,836, respectively. The results show that the purified protein is a homotetramer.

(A) SDS-PAGE analysis of samples taken during purification of the blue protein. Lane 1, molecular mass standards (numbers at the left are molecular masses in kDa); lane 2, (NH4)2SO4 fraction; lane 3, Q-Sepharose fraction; lane 4, Phenyl-sepharose fraction; lanes 5 and 6, hydroxyapatite fractions containing 3 and 6 μg of protein, respectively. (B) Native PAGE of purified blue protein. Lane 1, molecular mass standards; lanes 2 and 3, hydroxyapatite fraction containing 4 and 8 μg of protein, respectively. Proteins were stained with Coomassie Brilliant Blue R250
The ICP/MS analyses showed that the blue protein did not contain transition metals, and tests for glycosylation were negative (data not shown).
At pH 7.0, solutions of the blue protein showed absorbance maxima at 280, 378, and 633 nm. The color changed to green upon acidification to pH 3.0 with the appearance of new absorbance maxima at 360 and 670 nm with a shoulder at 703 nm (Fig. 3). The original spectrum and blue color was restored when the pH was adjusted back to 7.0. Extraction of the blue protein with cold 90% (v/v) acetone, followed by removal of the solvent, yielded a blue product, which was dissolved in methanol. The absorbance spectrum of the resulting solution showed maxima at 380 nm and a broad peak between 640 and 680 nm and was identical to an authentic sample of biliverdin IXα.

(A) Absorbance spectrum of purified blue protein (12.5 μg of protein/ml) in 50 mM Tris–HCl buffer, pH 7.0. (B) In 50 mM citrate buffer, pH 3.0
The amino acid composition of the blue protein is shown in Table 2, where it is compared to the amino acid compositions of butterfly, Pieris brassicae, bilin binding protein (BBP) and tobacco hornworm, Manduca sexta, insecticyanin. The blue protein differs from the two insecticyanins primarily in terms of the percentage of glutamic acid/glutamine, alanine, arginine, and tyrosine residues.
Attempts to determine the amino acid sequence of the blue protein were hindered due to a blocked N-terminus. In separate experiments, cyanogen bromide cleavage and endoproteinase digestion yielded seven internal peptides, which were separated by SDS-PAGE. The amino acid sequence of each peptide was determined and combination of overlapping sequences resulted in the identification of three different larger peptides. Standard NCBI Protein–Protein BLAST analysis of short peptide sequences showed the three peptides, designated A, B, and C (totaling 77 amino acids), to have the highest sequence identity to an unannotated protein, containing 181 amino acids, in the genome of the green spotted pufferfish, Tetraodon nigroviridis. The amino acid sequences of peptides A, B, and C are located at positions 24–53, 88–105, and 111–139, respectively (Fig. 4), and show 73% sequence identity to the T. nigroviridis protein. Table 4 shows alignments of the amino acid sequences of the three peptides with analogous regions in apolipoproteins D from zebrafish. Danio rerio (53%), guinea pig, Cavia porcellus (53% identity), mouse, Mus muscalus (49% identity), human, Homo sapiens (48% identity).

Alignments of amino acid sequences in peptides (A), (B), and (C) from the blue protein with predicted amino acid sequences at positions 24–53, 88–105, and 113–139, in an unnamed protein (NCBI Accession No. CAF98955) in the genome of the green spotted pufferfish, Tetraodon nigroviridis. Identical amino acids are shaded. Conservative amino acid substitutions are designated by +
Discussion
Several naturally occurring blue proteins have been described. They include copper proteins such as plastacyanin (Freeman and Guss 2001) and azurin (Kolczak et al. 2001); crustacyanin, a caretenoprotein complex found in the lobster, Homarus gammarus, carapace (Chayen et al. 2003); and BBPs found in insects (Riley et al. 1984; Huber et al. 1987; Saito 1998). The blue color in BBPs is due to the presence of biliverdin IXγ which is formed by the enzymatic cleavage of heme by heme oxygenase.
The blue protein purified in the present study is the first to be specifically associated with walleye. Initial studies showed the protein does not contain copper or any other transition metal. Its physical properties are different to those reported for crustacyanin. They are however, remarkably similar to several insect BBPs in terms of subunit composition, absorbance spectrum, and the presence of a biliverdin chromophore (Table 3). Although further studies are necessary to unequivocally establish that the chromophore is the biliverdin IXα isomer. These properties suggest that the blue protein is a biliprotein secreted by the fish into its skin mucus. We propose the name Sandercyanin for this new pigment.
The specific function of Sandercyanin is unknown. Blue walleye forms were known to occupy the deeper waters of the Great Lakes, leading Paradis and Magnan (2005) to suggest the blue color may provide an advantage to walleye in their search for forage fish. This seems reasonable in an environment that would be dominated by blue to blue–green light. A camouflage role has also been suggested for insect biliproteins when they are present in larval hemolymph. In this case the green color in the caterpillars is due to a combination of a biliprotein and a yellow carotenoid (Saito 1998). However, the amino acid sequences of the three sandercyanin internal peptides are surprisingly, similar to the predicted analogous sequences of an unannotated protein in the genome of the green spotted pufferfish and also to apolipoproteins D in zebra fish, guinea pig, mouse, and human (Table 4). These observations suggest a possible broader function for Sandercyanin. Apolipoproteins D are small secreted proteins which are characterized by their ability to bind and transport small hydrophobic molecules (Rassart et al. 2000). They, together with biliproteins, are members of the large lipocalin protein family (Flower 1996). The chromophore in Sandercyanin is a hydrophobic biliverdin derivative and transport of biliverdin to the mucus may be an indication of increased heme metabolism in blue walleye forms. In this context, it is of interest to note that chemical stress leads to increased biliverdin excretion in channel catfish, Ictalurus punctatus (van den Hurk 2006), although there was no evidence of pollution in the sites sampled in the present study. An alternative suggestion focuses on the ability of biliverdin to quench reactive oxygen radicals and the possible function of biliproteins in photoprotection (McDonagh 1979). Clearly, the physiological function of Sandercyanin awaits more detailed structure/function analyses. Such studies are ongoing in our laboratory.
Notes
www.bluewalleye.com.
References
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Chayen NE, Cianci M, Grossman JG et al (2003) Unravelling the structural chemistry of the colouration mechanism in lobster shell. Acta Crystallogr D Biol Crystallogr 59:2072–2082
Flower DR (1996) The lipocalin protein family: structure and function. Biochem J 318:1–14
Freeman HC, Guss JM (2001) Plastocyanin. In: Messerschmidt A, Huber R, Poulos T, Wieghardt K (eds) Handbook of metalloproteins. Wiley, Chichester, pp 1153–1169
Fujii R (1993) Colouration and chromatophores. In: Evans DH (ed) The physiology of fishes. CRC Marine Sciences Series, FL,USA, pp 535–562
Goodman WG, Adams B, Trost JT (1985) Purification and characterization of a biliverdin-associated protein from the hemolymph of Manduca sexta. Biochemistry 24:1168–1175
Huber R, Schneider M, Epp O et al (1987) Crystallization, crystal structure analysis and preliminary molecular model of the bilin binding protein from the insect Pieris brassicae. J Mol Biol 195:423–434
Kolczak U, Dennison C, Messerschmidt A et al (2001) Azurin and azurin mutants. In: Messerschmidt A, Huber R, Poulos T, Wieghardt K (eds) Handbook of metalloproteins. Wiley, Chichester, pp 1170–1194
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
McDonagh AF (1979) Bile pigments: bilatrienes and 5,15-biladienes. In: Dolphin D (ed) The porphyrins, vol 6. Academic, New York, pp 293–349
Paradis Y, Magnan P (2005) Phenotypic variation of walleye, Sander vitreus, in Canadian shield lakes: new insights on percid polymorphism. Environ Biol Fishes 73:357–366
Rassart E, Bedirian A, Do Carmo S et al (2000) Apolipoprotein D. Biochim Biophys Acta 1482:185–198
Regier H, Applegate VC, Ryder RA (1969) The ecology and management of walleye in western lake Erie. Great Lakes Fish Comm Tech Rep, AnnArbor, MI, p 101
Riley CT, Barbeau BK, Keim PS et al (1984) The covalent protein structure of insecticyanin, a blue biliprotein from the hemolymph of the tobacco hornworm, Manduca sexta L. J Biol Chem 259:13159–13165
Saito H (1998) Purification and properties of two blue biliproteins from the larval hemolymph and integument of Rhodinia fugax (Lepidoptera: Saturniidae). Insect Biochem Mol Biol 28:995–1005
Scott WB, Crossman EJ (1973) Freshwater fishes of Canada. Bulletin 184, Fisheries Research Board of Canada, Ottawa, p 966
Suter F, Kayser H, Zuber H (1988) The complete amino-acid sequence of the bilin-binding protein from Pieris brassicae and its similarity to the family of serum transport proteins like the retinol-binding proteins. Biol Chem Hoppe Seyler 369:497–505
van den Hurk P (2006) Bile fluorescence, heme oxygenase induction, and increased biliverdin excretion by mixtures of environmental toxicants. Aquat Toxicol 77:202–209
Yamanaka A, Ito T, Koga D et al (2000) Purification and characterization of biliverdin-binding protein from larval hemolymph of the swallowtail butterfly, Papilio xuthus L. Biosci Biotechnol Biochem 64:1978–1981
Acknowledgements
We thank Wensheng Liu for technical assistance, Lynn Teesch, Yalan Li, and Brian Morrison for help in MALDI-TOF/MS, amino acid sequence and amino acid composition analyses, Donald L. Simmons for transition metal analyses and Julie Nealson for help in preparation of the manuscript. Daniel Ferraro was supported by a NIH training grant in biotechnology (5 T32 GM08365-16).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yu, CL., Ferraro, D., Ramaswamy, S. et al. Purification and properties of Sandercyanin, a blue protein secreted in the mucus of blue forms of walleye, Sander vitreus . Environ Biol Fish 82, 51–58 (2008). https://doi.org/10.1007/s10641-007-9252-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10641-007-9252-3