
Summary
Background We assessed the safety, tolerability, and pharmacokinetics of mitochondrial complex 1 inhibitor ASP4132. Methods This phase I dose-escalation/dose-expansion study enrolled patients with treatment refractory advanced solid tumors to assess safety, dose-limiting toxicities (DLTs), efficacy and pharmacokinetic or oral ASP4132. Results Overall, 39 patients received ASP4132. Acceptable tolerability of ASP4132 5 mg in the first patient led to enrollment in the 10-mg dose cohort. After two DLTs at the 10-mg dose, additional patients were enrolled in the 5-mg cohort; a 7.5-mg cohort and two intermittent-dosing cohorts (ASP4132 10 mg for 3 days, then 4 days off; ASP4132 15 mg for 1 day, then 6 days off). ASP4132 5 mg was well tolerated; however, multiple DLTs such as fatigue, mental status changes, dizziness, lactic acidosis, enteritis, and posterior reversible encephalopathy syndrome were observed in higher dose cohorts (7.5-mg and intermittent 10-mg and 15-mg dose cohorts). Stable disease (+ 4 % to + 15 %) was observed in 8/39 (20.5 %) patients. ASP4132 plasma pharmacokinetics were characterized by high variability, with rapid absorption and accumulation from slow elimination. Conclusions ASP4132 showed limited clinical activity, and DLTs prohibited dose escalation. Further research is required to determine if DLTs will limit clinical activity of other mitochondrial complex I inhibitors. Clinical Trial ID (clinicaltrials.gov): NCT02383368, March 9, 2015.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Research has demonstrated that oxidative metabolism may be an important source of energy for tumor cells, suggesting that this pathway could potentially be exploited as an anticancer strategy. One such anticancer strategy is to make use of the inhibition of mitochondrial complex I, a critical step in oxidative phosphorylation. Mitochondrial complex I is the first step in the electron transport chain that binds and oxidizes NADH; subsequent transfer of the electrons results in formation of ATP [1]. Preclinical and epidemiological studies have given evidence in support of this concept, suggesting that the biguanides metformin and phenformin, mitochondrial complex I inhibitors, may have activity in cancer [2,3,4,5]. Indeed, metformin use in diabetic patients is associated with the reduced risk of cancer [6]. A number of other agents targeting mitochondrial complex I have been studied in preclinical models. For example, β-sitosterol prevented formation of brain metastases in a melanoma model [7], and papaverine is an effective radiosensitizer [8]. These various preclinical and clinical results suggest a potential role for mitochondrial complex I inhibitors in melanoma, prostate, breast, lung, ovarian, colorectal, and pancreatic cancers [7, 9, 10].
ASP4132 ditosilate is a novel mitochondrial complex I inhibitor formulated for oral administration. In vitro ASP4132 has demonstrated inhibition of cancer cell growth. In addition, regression and growth inhibition have been observed in in vivo animal models of cancer tumors (unpublished data). The objective of this phase I, first-in-human, open-label study of oral ASP4132 in patients with advanced and refractory malignancies was to determine the maximum tolerated dose, and to evaluate the safety, tolerability, and pharmacokinetics (PK) of ASP4132.
Materials and methods
Study design
This multicenter phase I study (clinicaltrials.gov identifier: NCT02383368) was designed to include a dose-escalation phase, followed by a dose-expansion phase in which patients received the maximum tolerated dose (MTD) or a lower dose recommended by the safety committee.
The planned sample size of the study allowed for enrollment of up to 100 patients. In the dose-escalation phase, sample size was based on the observed incidence of dose-limiting toxicities (DLTs); an accelerated 3 + 3 dose titration of cohorts of 3–6 patients per dose level was planned until the MTD was reached. The dose-evaluable set consisted of patients who received at least 80 % of the planned ASP4132 dose during a cycle, had sufficient safety evaluations, or experienced a DLT. The initial dose of ASP4132 was chosen based on animal toxicity studies. A monkey model was found to be the most sensitive species tested; this model was used to determine the most conservative human starting dose of 10 mg/day. In order to ensure patient safety, the initial dose was further reduced to 5 mg/day. A single patient was dosed with ASP4132 5 mg once-daily (Supplementary Fig. 1). After completion of one cycle with this patient, a second cohort of ASP4132 10 mg once-daily with 10 patients was opened. Due to tolerability concerns in the 10-mg once-daily cohort, the 5-mg once-daily cohort was re-opened and enrolled five patients; a 7.5-mg cohort was also enrolled. Subsequently, two additional intermittent dosing cohorts were enrolled: ASP4132 10 mg for 3 days on/4 days off, or 15 mg on one day followed by 6 days off.
ASP4132 was administered orally without food. Patients must not have eaten for at least 2 h before, and 1 h after, dosing. Similarly, concomitant medications were not to be administered within 2 h before or after dosing.
Patients
The study population consisted of patients with advanced solid tumors or lymphoma that were refractory to standard therapy. For the dose-escalation phase, patients with refractory advanced breast cancer, non-small cell lung cancer (NSCLC), malignant melanoma, colorectal cancer, ovarian cancer, castration resistant prostate cancer, or lymphoma were included, as these tumors are known to be dependent on oxidative phosphorylation [7, 9, 10]. Patients with other cancers could be considered for inclusion after discussion with the sponsor/medical monitor.
Patients were eligible for study enrollment if they had a life expectancy of ≥ 3 months and an Eastern Cooperative Oncology Group (ECOG) performance status ≤ 2. Patients who had undergone radiotherapy or surgery within 2 weeks prior to treatment with ASP4132, patients who had not discontinued all previous systemic cancer therapies at least 14 days prior to treatment, or had not fully recovered from acute toxicities of therapy (except alopecia), were excluded. Additional detailed exclusion criteria can be found in the study protocol included in Supplementary Information.
Assessments
Safety was assessed through vital signs, weight, laboratory assessments, physical examinations, electrocardiogram, echocardiogram or multigated acquisition scan, ophthalmic examinations, and reporting of adverse events (AEs) as graded by the National Cancer Institute’s Common Terminology Criteria for Adverse Events v4.03 (NCI-CTCAE). The grading of lactatemia is shown in Supplementary Table 1.
DLTs were defined as febrile neutropenia, asymptomatic grade 4 neutropenia or thrombocytopenia, or grade 3 thrombocytopenia with bleeding, or any grade ≥ 3 AE (except grade 3 nausea, vomiting or diarrhea that was managed to grade ≤ 1 with standard antiemetic or antidiarrheal medications, or grade 3 asthenia for < 7 days), lactic acidosis defined as arterial pH < 7.35 or grade 3 acidosis in the presence of ≥ 5 mmol lactate, or ≥ 14 days continuous delay in ASP4132 due to hematologic or nonhematologic toxicity. The safety analysis set included all patients who received at least one dose of study drug.
Prior to Cycle 1, a single dose of ASP4132 was administered on Day − 4, and samples for PK analysis were obtained immediately prior to dosing, and 0.5, 1, 2, 3, 4, 6, 8, 24, 48, 72, and 96 h after dosing. Samples were also drawn prior to dosing during Cycle 1 on Days 8, 15, 22, and 28; 2 h after dosing on Day 15; and 0.5, 1, 2, 3, 6, 8, and 24 h after dosing on Day 28. A sample was also taken immediately prior to dosing on Day 15 of Cycle 2. Plasma concentration data were used to estimate standard PK parameters following single and multiple doses. The PK analysis set consisted of the subset of patients for whom sufficient plasma concentration data were available to facilitate derivation of at least one PK parameter.
Efficacy assessments were objective response rate (complete response [CR] plus partial response [PR]), disease control rate (CR + PR + stable disease [SD]), duration of response, and progression-free survival. Response was assessed using Response Evaluation Criteria in Solid Tumors (RECIST) v.1.1 criteria for tumors to which these criteria apply; for other tumor types, indication specific response criteria were used.
Results
Baseline characteristics
A total of 39 patients were dosed with ASP4132. The majority of patients enrolled in the study were female (77 %) and white (92 %), and ranged in age from 24 to 80 years (mean 59 years) (Table 1). The most common cancer diagnoses for patients enrolled in the trial were breast cancer, colorectal cancer, and NSCLC.
Safety
The first patient enrolled in the initial ASP4132 5-mg cohort experienced no treatment-related toxicities during the DLT assessment period, leading to enrollment of the 10-mg dose cohort. However, ASP4132 10 mg was not well tolerated at this dose, with two DLTs observed; one patient had grade 4 mental status changes, and one patient had grade 3 fatigue. As a result, additional patients were enrolled in the 5-mg dose cohort and a 7.5-mg once-daily dose cohort was also enrolled. As with the initial patient, ASP4132 5 mg was well tolerated and there were no DLTs. The ASP4132 7.5-mg dose was poorly tolerated and two additional DLTs were reported; one patient developed lactic acidosis and dizziness, and one patient experienced grade 3 fatigue.
Based on the safety results observed with once-daily dosing, PK results suggesting significant drug accumulation (see Pharmacokinetics and Pharmacodynamics below), as well as efficacy and PK modeling data from a preclinical in vivo experiment using an MD-MB-453 breast cancer xenograft model (data not shown), patients were enrolled into two unplanned intermittent-dose cohorts. For the first intermittent-dose cohort, the ASP4132 dose was 10 mg/day for 3 days followed by 4 days off drug; for the second intermittent-dose cohort, the ASP4132 dose was 15 mg weekly on one day followed by 6 days off drug. Both intermittent-dosing regimens were poorly tolerated and there were one and two DLTs in the 10-mg and 15-mg treatment cohorts, respectively. The patient in the ASP4132 10-mg intermittent cohort with a DLT developed grade 3 enteritis over a period of 8 days. In the ASP4132 15-mg intermittent-dosing cohort, one patient developed posterior reversible encephalopathy syndrome over a period of 4 days, and one patient experienced grade 3 mental status changes. There were three deaths during the study, but none were considered related to ASP4132.
Treatment-emergent AEs (TEAEs) were reported in all patients enrolled in this study (Supplementary Table 2). The most common TEAEs were nausea (67 %), vomiting (51 %), fatigue (46 %), increased blood lactic acid (36 %), and decreased appetite (31 %). Treatment-related AEs occurred in 35 of 39 patients (90 %) (Table 2). The most frequent of these were nausea (62 %), vomiting (41 %), increased blood lactic acid (33 %), and fatigue (31 %).
Pharmacokinetics and pharmacodynamics
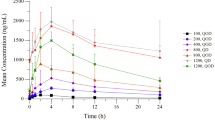
Three patients vomited shortly after dosing during the initial single dose PK phase and were omitted from the analysis. In addition, sampling was stopped prior to 96 h in a number of patients; as a result, the area under the curve from 0 to 24 h and 0–96 h (AUC24 and AUC96) could not be calculated for all patients. ASP4132 was absorbed rapidly and the time to peak concentration (tmax) appeared to be independent of dose (Fig. 1a, Supplementary Table 3). There was a tendency toward slower absorption after multiple dosings, but this was not consistent across dose cohorts and may reflect the small patient numbers and wide variability between patients (Fig. 1b and c). Plasma concentrations of ASP4132 decreased slowly after reaching peak concentration (Cmax). Plasma concentration profiles were often irregular after reaching Cmax, with multiple peaks appearing, making it difficult to calculate half-life (t1/2) and other t1/2-dependent parameters, including AUCinf, apparent total systemic clearance (CL/F), and apparent volume of distribution (Vz/F).

Plasma concentration profile of ASP4132. a Single oral dose (Cycle 1 Day − 4). b Multiple dosing in once-daily cohorts (Cycle 1 Day 28).c Multiple dosing in intermittent cohorts (Cycle 2 Day 1). Abbreviations: INT, intermittent dosing; QD, once daily
After a single dose of ASP4132, AUC96 and Cmax increased dose proportionally, but with large 90 % confidence intervals for the slope. No such relationship was observed following multiple dosing, but this may merely reflect the limited available data.
It is not clear from these results whether steady-state concentrations of ASP4132 were reached because minimum concentration (Ctrough) values suggested that further increases with continued dosing were possible, indicating a slow elimination of the drug from circulation. Consistent with this finding, high accumulation ratio values indicated extensive accumulation of ASP4132.
Serum lactate concentrations revealed no clear dose dependency or change over time (Supplementary Table 4). Given the high PK variability of ASP4132, an exposure-response analysis was performed with all available data. The baseline lactate concentration (standard deviation) was 1.25 (0.12) mmol/L, and the ASP4132 concentration required to yield a 50 % increase at any timepoint was 52.1 (25.7) ng/mL. There was a plateau in serum lactate levels with increasing ASP4132 concentrations above 40 ng/mL; the estimated maximum lactate concentration reached in response to ASP4132 treatment was 3.83 (1.38) mmol/L (Supplementary Fig. 2).
Efficacy
No objective responses (CRs or PRs) were observed at any dose of ASP4132 (Table 3). Progressive disease was observed in 16 patients (41 %) and 14 patients (36 %) were not evaluable. Stable disease ranging from + 4 % to + 15 % change in sum of target lesions was observed in 8/39 patients for a disease control rate of 21 %. All but one patient with SD progressed or withdrew due to AEs before 6 months; one female patient with cervical cancer who received ASP4132 5 mg sustained SD during 2 years (730 days) of treatment, before experiencing progressive disease at Day 787 (Fig. 2). Because of the lack of objective responses and poor tolerability of ASP4132 at doses higher than 5 mg once-daily, the dose-expansion phase of this study was not opened and the study was terminated.

Swimmer plot of patient disposition. Gray bars depict total exposure to ASP4132 for each patient in days. Symbols indicate reasons for discontinuation and/or disease status at the time of discontinuation. For patients in whom SD was observed, interim assessments before discontinuation are also shown. Abbreviations: AE, adverse event, DLT, dose-limiting toxicity; INT, intermittent dosing; PD, progressive disease; SD, stable disease
Discussion
Patients treated with ASP4132 displayed DLTs at doses lower than were anticipated to show clinical efficacy. This therefore precluded escalation to doses that would be associated with clinical activity. The occurrence of DLTs in patients treated with ASP4132 10 mg/day precluded further evaluation of the daily dosing schedule. As a result, unplanned intermittent-dosing cohorts were added to this study in an attempt to achieve efficacious drug exposure while mitigating the observed toxicity. Unfortunately, although the rate of DLTs in the intermittent-dosing cohorts did not prohibit further dose escalation, the nature of the events that did occur (grade 4 posterior reversible encephalopathy syndrome and grade 3 mental status changes) prohibited further dose escalation. Most other reported AEs were those typical of chemotherapy, and included nausea, vomiting, fatigue, and decreased appetite. At doses higher than 5 mg, plasma concentrations of ASP4132 did not reach levels associated with therapeutic efficacy in preclinical models and no objective responses were demonstrated.
Pharmacokinetic data indicated that the drug exposure attained with oral dosing of ASP4132 was lower than that predicted to have efficacy based on mouse xenograft model data (data not shown). This was reflected in the lack of an objective response in any patient in this study. However, there was some suggestion of clinical activity in 21 % of patients who experienced SD, including one patient in the 5-mg once-daily cohort who had SD for 2 years before disease progression. The PK of ASP4132 was characterized by a rapid increase in plasma concentration to reach Cmax, followed by slow elimination and substantial accumulation. A full characterization of the PK of ASP4132 was complicated by the appearance of multiple peaks in serum plasma concentration after reaching Cmax. The long half-life and drug accumulation may in part explain the toxicity of ASP4132, especially with once-daily dosing. This PK profile made it difficult to design alternative dosing schedules that may mitigate the TEAEs after ASP4132 treatment to reach serum plasma concentrations associated with clinical activity.
Despite the limited clinical activity of ASP4132, pharmacodynamic data, as well as the observed toxicity, indicated that the target mitochondrial complex I was engaged in this study. Increased serum lactate was reported as an AE in one-third of patients in this study, and two of 39 patients experienced lactic acidosis, suggesting that glycolysis was upregulated following inhibition of the mitochondrial complex I with ASP4132. An increase in serum lactate is expected in tumor cells following inhibition of the mitochondrial complex I, as anaerobic glycolytic activity is increased to compensate for the lack of energy production in oxidative phosphorylation [4]. It is believed that this upregulation of glycolysis during a lack of energy production via oxidative phosphorylation is triggered through activation of adenosine monophosphate kinase, which in turn stimulates glycolysis through activation of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 2/3 [11, 12]. This increased glycolytic activity leads to increased production of pyruvate and lactate, and an increased risk of lactic acidosis [4]. Although serum lactate levels of ASP4132 did not show any clear dose-dependency in this study, there was an ASP4132 plasma concentration-dependent increase in lactate, with a plateau above 40 ng/mL.
This switch to glycolysis has been posited as a reason for lack of activity of other small molecule mitochondrial complex I inhibitors. In a chronic lymphocytic leukemia mouse model, the lack of activity of IACS-01075 was attributed to this switch as an adaptive mechanism; the authors of an article examining the study suggested that simultaneous use of a glycolysis inhibitor may be required to demonstrate efficacy [13]. Because oxidative phosphorylation is essential for all cellular functions in the body, its inhibition and subsequent switch to glycolysis would be expected to have a broad range of side effects. The observed increase in serum lactate, and the fact that this indicates a pharmacodynamic effect of ASP4132, therefore provides a possible explanation for the observed toxicity. Preliminary results of first-in-human studies of two other mitochondrial complex I inhibitors have recently been presented. IACS-010759 administered at the MTD did demonstrate some preliminary activity: one of five patients achieved a PR [14]. IM156 has been well tolerated to date, but the MTD has not been established and, similar to our study, the best clinical activity demonstrated to date is SD [15]. Additional clinical research will be required to determine if these agents can deliver meaningful clinical activity at tolerable doses, either alone or in combination with agents with different mechanisms of action.
In summary, this study demonstrated that ASP4132 successfully engaged its target, as demonstrated by changes in serum lactate. However, ASP4132 demonstrated only limited clinical activity (20.5 % of patients achieved SD) and was associated with significant toxicity, including DLTs and mechanism-related toxicities. As a result, the study was terminated early and clinical development of ASP4132 was halted. While it is not clear whether DLTs will preclude the clinical development of other mitochondrial complex I inhibitors, further preclinical investigations may be required to shed more light on the feasibility of targeting the mitochondrial complex.
References
Mimaki M, Wang X, McKenzie M, Thorburn DR, Ryan MT (2012) Understanding mitochondrial complex I assembly in health and disease. Biochim Biophys Acta 1817(6):851–862. https://doi.org/10.1016/j.bbabio.2011.08.010
Bridges HR, Jones AJ, Pollak MN, Hirst J (2014) Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem J 462(3):475–487. https://doi.org/10.1042/BJ20140620
Pollak MN (2012) Investigating metformin for cancer prevention and treatment: the end of the beginning. Cancer Discov 2(9):778–790. https://doi.org/10.1158/2159-8290.CD-12-0263
Owen MR, Doran E, Halestrap AP (2000) Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J 348 Pt 3:607–614
Rajeshkumar NV, Yabuuchi S, Pai SG, De Oliveira E, Kamphorst JJ, Rabinowitz JD, Tejero H, Al-Shahrour F, Hidalgo M, Maitra A, Dang CV (2017) Treatment of pancreatic cancer patient-derived xenograft panel with metabolic inhibitors reveals efficacy of phenformin. Clin Cancer Res 23(18):5639–5647. https://doi.org/10.1158/1078-0432.CCR-17-1115
Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD (2005) Metformin and reduced risk of cancer in diabetic patients. BMJ 330(7503):1304–1305. https://doi.org/10.1136/bmj.38415.708634.F7
Sundstrom T, Prestegarden L, Azuaje F, Aasen SN, Rosland GV, Varughese JK, Bahador M, Bernatz S, Braun Y, Harter PN, Skaftnesmo KO, Ingham ES, Mahakian LM, Tam S, Tepper CG, Petersen K, Ferrara KW, Tronstad KJ, Lund-Johansen M, Beschorner R, Bjerkvig R, Thorsen F (2019) Inhibition of mitochondrial respiration prevents BRAF-mutant melanoma brain metastasis. Acta Neuropathol Commun 7(1):55. https://doi.org/10.1186/s40478-019-0712-8
Benej M, Hong X, Vibhute S, Scott S, Wu J, Graves E, Le QT, Koong AC, Giaccia AJ, Yu B, Chen CS, Papandreou I, Denko NC (2018) Papaverine and its derivatives radiosensitize solid tumors by inhibiting mitochondrial metabolism. Proc Natl Acad Sci U S A 115(42):10756–10761. https://doi.org/10.1073/pnas.1808945115
Cerezo M, Tichet M, Abbe P, Ohanna M, Lehraiki A, Rouaud F, Allegra M, Giacchero D, Bahadoran P, Bertolotto C, Tartare-Deckert S, Ballotti R, Rocchi S (2013) Metformin blocks melanoma invasion and metastasis development in AMPK/p53-dependent manner. Mol Cancer Ther 12(8):1605–1615. https://doi.org/10.1158/1535-7163.MCT-12-1226-T
Brown KA, Samarajeewa NU, Simpson ER (2013) Endocrine-related cancers and the role of AMPK. Mol Cell Endocrinol 366(2):170–179. https://doi.org/10.1016/j.mce.2012.06.016
Jeon SM (2016) Regulation and function of AMPK in physiology and diseases. Exp Mol Med 48(7):e245. https://doi.org/10.1038/emm.2016.81
Gotlieb WH, Saumet J, Beauchamp MC, Gu J, Lau S, Pollak MN, Bruchim I (2008) In vitro metformin anti-neoplastic activity in epithelial ovarian cancer. Gynecol Oncol 110(2):246–250. https://doi.org/10.1016/j.ygyno.2008.04.008
Vangapandu HV, Alston B, Morse J, Ayres ML, Wierda WG, Keating MJ, Marszalek JR, Gandhi V (2018) Biological and metabolic effects of IACS-010759, an OxPhos inhibitor, on chronic lymphocytic leukemia cells. Oncotarget 9(38):24980–24991. https://doi.org/10.18632/oncotarget.25166
Yap TA, Ahnert JR, Piha-Paul SA, Fu S, Janku F, Karp DD, Naing A, Dumbrava EEI, Pant S, Subbiah V, Tsimberidou AM, Hong DS, Rose KM, Xu Q, Vellano CP, Mahendra M, Jones P, Di Francesco ME, Marszalek JR, Meric-Bernstam F (2019) Phase I trial of IACS-010759 (IACS), a potent, selective inhibitor of complex I of the mitochondrial electron transport chain, in patients (pts) with advanced solid tumors. J Clin Oncol 37(15_suppl):3014
Rha SY, Beom SHB, Kim GM, Shin YG, Yim D-S, Kim HS, Chang JH, Cheong J-H, Lee YW, Chong YS, O’Neill V, Yoo SH, Janku, F (2019) Phase I study of an oxidative phosphorylation inhibitor IM156 in patients with advaned solid tumors. In: American Associaiton for Cancer Research, Atlanta, p CT021/016
Acknowledgements
Medical writing/editorial support was provided by James Street, PhD, and Elizabeth Hermans, PhD, of OPEN Health Medical Communications, Chicago, IL, and funded by the study sponsor.
Funding
This work was supported by Astellas Pharma, Inc.
Author information
Authors and Affiliations
Contributions
FJ, PL, ASM, TW, AMB, and HAB designed the research study; FJ, RN, AMB, and HAB developed the methodology; FJ, PL, ASM, RN, AS, and AMB acquired the data; FJ, PL, ASM, RN, AS, TW, AMB, JS, and HAB analyzed and interpreted the data; FJ, PL, ASM, RN, AS, TW, AMB, JS, and HAB critically reviewed and revised the manuscript; FJ, PL, RN, and AS provided administrative, technical, or material support; FJ, PL, ASM, RN, AS, AMB, and HAB supervised the study. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Dr Filip Janku reports grants from Novartis, Genentech, BioMed Valley Discoveries, Plexxikon, Deciphera, Piqur, Symphogen, Bayer, FujiFilm Corporation, Astex, Asana, Astellas, Agios, Proximagen, and Bristol-Myers Squibb; and other support from Deciphera, IFM Therapeutics, Synlogic, Guardant Health, Ideaya, PureTech Health, Jazz Pharmaceuticals, Trovagene, Immunomet, Primmune Therapeutics, and Sotio, outside the submitted work.Dr Patricia LoRusso reports honorarium from advisory boards from AbbVie, Agios, GenMab, Genentech, CytomX, Takeda, Cyrexa, Agenus, IQVIA, TRIGR, Pfizer, I-MAB, ImmunoMet, Black Diamnod, Glaxo-Smith Kline, QED Therapeutics, AstraZeneca, EMD Serono, Shattuck, Astellas, Salarius, Silverback, MacroGenics; and has been a consultant for Five Prime, Halozyme, Roche-Genentech, SOTIO, and Tyme.Dr Aaron Mansfield has no direct conflicts, but reports research support from Bristol-Meyers Squibb, Novartis, and Verily, remuneration to his institution for participation on advisory boards for AbbVie, Astra Zeneca, BMS, and Genentech, and is a non-remunerated director of the Mesothelioma Applied Research Foundation. Dr Rita Nanda reports personal fees from Aduro, grants and personal fees from AstraZeneca, personal fees from Athenex, grants and personal fees from Celgene, personal fees from Daiichi Sankyo, grants and personal fees from Genentech, personal fees from Macrogenics, grants and personal fees from Merck, grants and personal fees from Novartis, grants and personal fees from Pfizer, personal fees from Puma, personal fees and other from G1 Therapeutics, other from OBI Pharma, grants from Immunomedics, grants from Odonate, and grants from Seattle Genetics, outside the submitted work. Dr Alexander Spira reports grants and non-financial support from Astellas during the conduct of the study; grants, personal fees, and non-financial support from Cytomx; grants, personal fees, and non-financial support from AstraZeneca; and non-financial support from Roche, outside the submitted work.
Dr Tianli Wang has nothing to disclose. Dr Amal Melhem-Bertrandt was an employee of Astellas at the time of the study. Jennifer Sugg is an employee of Astellas and holds stock in AstraZeneca. Dr Howard A. Ball has nothing to disclose.
Ethics approval
The study was conducted in accordance with Good Clinical Practice, the International Council for Harmonisation, and guidelines governing clinical study conduct and the ethical principles originating from the Declaration of Helsinki.The study was approved by the respective Institutional Review Boards.
Consent to participate
Written informed consent was obtained from all individual participants included in the study.
Consent for publication
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
ESM 1
(PDF 319 KB)
Rights and permissions
About this article

Cite this article
Janku, F., LoRusso, P., Mansfield, A.S. et al. First-in-human evaluation of the novel mitochondrial complex I inhibitor ASP4132 for treatment of cancer. Invest New Drugs 39, 1348–1356 (2021). https://doi.org/10.1007/s10637-021-01112-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-021-01112-7