
Summary
Background Dabrafenib is a BRAF inhibitor that has demonstrated clinical activity with a good tolerability profile in patients with BRAF V600E mutated metastatic melanoma. This study evaluated the safety and tolerability, pharmacokinetics and preliminary efficacy of dabrafenib in Japanese patients. Methods This phase I, open-label, dose escalation study was conducted in 12 Japanese patients with BRAF V600 mutation positive solid tumours. Primary endpoint was safety, assessed by monitoring and recording of all adverse events (AEs), serious AEs, drug-related AEs; secondary endpoints were pharmacokinetic profiles and efficacy measured by tumour response. This study is registered with ClinicalTrials.gov, number NCT01582997. Results Of the 12 patients enrolled, 3 each received 75 mg and 100 mg dabrafenib while 6 received 150 mg dabrafenib twice daily orally. Melanoma and thyroid cancer were the primary tumours reported in 11 (92%) and 1 (8%) patients respectively. Most AEs were grade 1 or 2 and considered related to study treatment. Most common AEs reported in the 12 patients were alopecia in 7 (58%); pyrexia, arthralgia and leukopenia in 6 (50%) each, hyperkeratosis and nausea in 4 (33%) each. Partial response as best overall response was reported in 7 of 12 (58%) patients and in 6 (55%) with malignant melanoma. No dose-limiting toxicity (DLTs) were reported during the DLT evaluation periods. Conclusions Dabrafenib was well tolerated and rapidly absorbed administered as single- or multiple dose. Comparable safety and pharmacokinetic profiles were observed compared with non-Japanese patients. Dabrafenib has promising clinical activity in Japanese patients with BRAF mutated malignant melanoma.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
v-raf murine sarcoma viral oncogene homolog B1 (BRAF) is a serine/threonine protein kinase that is activated by somatic point mutations in human cancer. BRAF is a key molecule of the rat sarcoma gene (RAS) that activates the mitogen-activated protein (MAP) kinase/extracellular signal-regulated kinase (ERK) signaling pathway leading to increased cell growth [1]. Mutations in the BRAF gene have been identified in approximately 7% of all cancers [1], including 60–70% of melanomas, 15% of papillary thyroid carcinomas [2], and 12% of colorectal cancers [3]. BRAF mutations have also been detected to a lesser extent in of 1.6–4.9% of non-small-cell lung cancers and in almost all patients with hairy cell leukemia [4] and papillary pharyngeal cancer [5].
Several genetic mutations have been shown to contribute to the development and progression of melanomas. Approximately 50% of cutaneous melanoma cases have activating mutations in BRAF [1], wherein BRAF mutations are common in melanomas that arise without chronic sun-induced damage and are rare in melanomas arising from mucosal or acral sites [6]. Of the observed BRAF mutations in melanomas, >90% are single nucleotide mutations due to a substitution of glutamic acid for valine at codon 600 (BRAF V600E: nucleotide 1799 thymidine > adenosine) at nucleotide 1799 in the BRAF gene and less commonly due to substitution with lysine (BRAF V600K) [1]. The substitution leads to elevated BRAF levels that further stimulate the ERK pathway, leading to cancer formations [7]. BRAF V600E/K has been implicated in different mechanisms of melanoma progression and activation of the downstream MEK/ERK pathway [8, 9].
BRAF V600 mutations have been observed in Japanese patients with the similar types of cancers, including 30.4% of malignant melanoma [10], 28–53% of papillary thyroid cancers [11,12,13], 1–6.5% of colorectal cancers [14, 15], and 9% of ovarian cancers [16]. There is increased understanding of the carcinogenic role of BRAF, and genetic tests can determine the presence of BRAF mutations, which can form the basis of a novel and promising therapy.
With the identification of the important role that BRAF mutations play in melanoma, the recent focus of research has been to develop selective BRAF inhibitors for the treatment of malignant melanoma. Clinical trials with nonselective or type 2 BRAF inhibitors such as Sorafenib [17, 18], RAF256 [19] did not demonstrate clinical efficacy when given as monotherapy and this was thought to be due to the nonselective nature of the inhibition. Furthermore, significant toxicity was also reported. Selective or type 1 BRAF inhibitors bind the active conformation of BRAF kinase and have demonstrated promising results in clinical trials with drugs such as vemurafenib [20, 21] and dabrafenib [22,23,24].
Dabrafenib is a potent adenosine triphosphate (ATP) competitive inhibitor of BRAF kinase, selective for the BRAF V600E/K mutation in kinase screening panels, cell lines, and xenografts [25]. Furthermore, dabrafenib demonstrated suppression of a downstream pharmacodynamic biomarker (phosphorylated ERK [pERK]) in tumor cell lines, showed anti-proliferative activity against multiple BRAF-mutant tumor cell lines, and achieved biomarker suppression and tumor regression in BRAF-mutant xenograft models. Dabrafenib is approved in the USA and Europe for the treatment of unresectable or metastatic BRAF V600E-positive melanoma. This phase 1 trial was conducted to investigate the safety, tolerability, pharmacokinetics (PK), and efficacy of dabrafenib in Japanese patients with BRAF V600E/K mutation-positive advanced solid tumors.
Materials and methods
Patient eligibility
Men and women (of non-childbearing potential or childbearing potential with a negative serum pregnancy test 7 days prior to the first dose of medication and using adequate contraception until 4 weeks after the last dose of study medication) were included in the study. The patients had to be aged ≥20 years with a histologically or cytologically confirmed diagnosis of BRAF V600E/K mutation-positive advanced solid tumor not responsive to standard therapy or for which there was no approved or curative therapy. Other inclusion criteria included patients negative for hepatitis B or C virus test, and had an Eastern Cooperative Oncology Group performance status (ECOG-PS) score of 0 or 1, and had adequate organ functions. Patients were excluded if they had a history of other malignancy within the past 5 years; were required to receive concomitant cancer therapy; had received treatment with an investigational anti-cancer drug within 28 days or its 5 half-lives, whichever was longer, preceding the first dose of the study drug; had received prior treatment with a BRAF or MEK inhibitor; had a history of acute coronary syndromes, coronary angioplasty, or stenting within the past 24 weeks, had QTc interval ≥ 480 msecs; had grade 2 or greater valvular heart disease as defined by the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) v4.0; or had Class II, III, or IV heart failure, as defined by the New York Heart Association (NYHA) functional classification system.
Study design
This phase I, open-label, dose-escalation study was conducted in Japanese patients with BRAF V600 mutation-positive solid tumors. All patients provided written informed consent before participating in any study procedures. The study was conducted in accordance with the International Conference on Harmonization (ICH) Guidelines for Good Clinical Practice (GCP) and the Declaration of Helsinki. All protocols and amendments were approved by the independent ethics committee or institutional review board for each study center. This trial was registered with ClinicalTrials.gov (NCT01582997).
Patients were tested for BRAF V600E/K mutations prior to treatment to determine study eligibility. Tumor BRAF mutation was tested using a direct sequencing method, and all baseline lesion assessments were performed within 28 days prior to start of the study treatment. Enrolled patients received a starting dose of dabrafenib 150 mg orally daily (75 mg twice daily [BID]), and dose escalation was conducted to assess the safety, tolerability, single- and multiple-dose PK profile, and preliminary efficacy of dabrafenib. A single dose of dabrafenib was administered on Day −7, and not administered until Day −1 for PK blood sampling until 168 h after the dose. A continuous daily dosing schedule was started from Day 1 until Week 12 (Fig. 1). The dose levels evaluated in the study included 75 mg, 100 mg, and 150 mg according to a BID dosing schedule. Patients were treated with dabrafenib until disease progression, or an unacceptable adverse event occurred or death. A dose escalation decision was made after safety assessment for dose-limiting toxicity (DLT) was determined according to a standard 3 + 3 dose-escalation design. The DLT evaluation period was defined as the period from the first 28 days after administration of the first dose (i.e., during 7 days after a single administration and 21 days after starting continuous BID administration). The key DLT criteria were as follows: grade 4 hematologic toxicity; grade 3 or 4 non-hematologic toxicity and rash, nausea, vomiting, and diarrhea, only if controlled with supportive therapy); rash grade 3 or greater that required dose reduction despite supportive care; grade 2 or greater non-hematologic toxicity that was considered dose limiting in the judgment of the investigator; treatment delay of greater than 14 consecutive days due to unresolved toxicity; and any new grade 2 or greater valvular heart disease as defined by the NCI CTCAE v4.0; patients with significant alteration in cardiac valve morphology from baseline.

Study design. DLT, dose limiting toxicity, PK, pharmacokinetics
Study assessments
Safety, the primary assessment, included monitoring and recording of all adverse events (AEs), serious adverse events (SAEs), drug-related AEs, discontinuations, and other notable laboratory abnormalities. Physical examination, measurement of vital signs, electrocardiography, echocardiography and monitoring of hematology and blood chemistry were performed at regular intervals during the study period. AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA v18.0), grouped by system organ class and preferred term, and were graded according to the NCI CTCAE v4.0. Secondary assessments included efficacy, which was measured by tumor response as defined by Response Evaluation Criteria in Solid Tumors (RECIST) v1.1. Other secondary endpoints included PK assessment of dabrafenib and its metabolites (GSK2285403 (hydroxylated metabolite), GSK2298683 (carboxylated metabolite), and GSK2167542 (demethylated metabolite) following single and multiple dosing, which included area under the curve (AUC), maximum plasma concentration of the drug (Cmax), time to reach maximum concentration (Tmax), terminal half-life and/or effective half-life (t1/2) and clearance following oral dosing (CL/F)). Full PK sampling was performed from Day −7 and Day 21, and trough PK sampling was performed every 3 weeks from Week 6 until Week 15 (Sampling points: Day 1, Day 8, Day 15, Week 6, Week 9, Week 12 and Week 15, and at pharmacodynamic observation). Plasma concentrations of dabrafenib and its metabolites were determined using approved analytical methodology.
Statistical analyses
Sample size
Eighteen patients were the maximum number planned to be enrolled, based on pre-defined criteria for dose selection but not driven by statistical considerations. The primary safety and efficacy analyses were performed in the “all treated patients” (ATS) population, which comprised all patients who received the study drug at least once. All patients who completed DLT assessments appropriately were included in the DLT assessment population, and all patients for whom a PK sample was obtained and analyzed comprised the PK population. The lesion data were listed for each patient with solid tumors. Overall response rate, defined as the percentage of patients who had a confirmed complete response (CR) or partial response (PR), was estimated along with exact 95% confidence intervals (CIs). For the safety analyses, all DLTs were recorded and listed, and AEs, drug-related AEs, SAEs, and AEs leading to discontinuation were reported as summaries; all summaries included data from scheduled assessments only. For the efficacy analyses, anti-tumor activities were calculated based on clinical evidence and RECIST v1.1 criteria for solid tumors. Response, measured as CR, PR, stable disease (SD), and progressive disease (PD), was listed and summarized based on the dose cohort. The PK analyses of dabrafenib and its metabolites were performed using non-compartmental analysis, and the PK parameters were listed and summarized descriptively by dose cohort.
Results
Patient disposition and baseline characteristics
A total of 12 patients were enrolled in the study and received dabrafenib 75 mg BID (n = 3), dabrafenib 100 mg BID (n = 3), and dabrafenib 150 mg BID (n = 6) at 2 study centers between May 2012 and April 2015. All 12 patients completed the DLT evaluation period and continued the study treatment after the DLT evaluation period. At the time of the final analysis, all patients discontinued the study treatment due to disease progression and completed study evaluation. Baseline characteristics of the patients enrolled in the study are summarized in Table 1.
The median duration (range) of exposure to dabrafenib was 372.0 (314–402) days in the 75 mg cohort, 253.0 (129–629) days in the 100 mg cohort, and 124.5 (19–669) days in the 150 mg cohort. An equal number of men and women were included in the study, with a median age at screening of 47.5 years. The major types of primary tumors were melanomas in 11 (92%) patients and thyroid cancer in 1 (8%) patient. The histological types of melanoma were nodular melanoma (17%), malignant melanoma not otherwise specified (NOS; 8%), superficial spreading melanoma (8%) and other (8%). The histological types of melanoma were unknown seen in half of the patients (50%). One patient (8%) was detected with papillary adenocarcinoma of the thyroid.
At the time of screening, 10 (83%) patients had stage IV disease. All patients had BRAF V600E mutation-positive tumors, detected by using a direct sequencing method. All patients received prior anti-cancer therapy. Chemotherapy was taken by 3 patients in the dabrafenib 75 mg cohort, 1 patient in the 100 mg cohort, and 6 patients in the 150 mg cohort. All 3 patients from the dabrafenib 75 mg and 100 mg cohorts, respectively, and 5 out of 6 patients from the 150 mg cohort had undergone prior cancer-related surgical procedures.
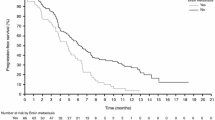
As the best overall response, PR in 7 (58%) out of 12 patients, 1 with thyroid cancer and 6 with melanoma. Of the 11 patients with melanoma, 6 (55%) had a tumor response. Overall, 2 (17%) patients with melanoma had SD and 3 (25%) patients with melanoma had PD. Table 2 depicts the overall best response seen on exposure to dabrafenib per dose cohort.
Safety findings
A summary of all AEs in the study population classified as per system organ class and preferred term is presented in Table 3. No DLTs were reported during the DLT evaluation periods. Grade 3 laboratory abnormalities were observed in 2 patients in the dabrafenib 150 mg cohort, which were not considered as AEs by the investigators. AEs and drug-related AEs were reported in all 12 patients who received study treatments. The AEs reported in a minimum of 4 patients out of the 12 were alopecia in 7 (58%); pyrexia, arthralgia, and leukopenia in 6 (50%) each; and hyperkeratosis and nausea in 4 (33%) each. Of these, alopecia (n = 6 [50%]), pyrexia (n = 6 [50%] patients), arthralgia (n = 6 [50%] patients), leukopenia (n = 5 [42%] patients), and hyperkeratosis (n = 4 [33%] patients) were regarded as drug-related AEs and graded as grade 1 or 2. Grade 1 AEs were observed in 2 patients and grade 2 AEs in 9. AEs classified as grade 3 were observed in 1 patient (pain and lymphopenia) and were not considered to be related to the study treatment but to primary cancer. No grade 4 or 5 AEs were observed in any of the cohorts. Table 4 summarizes the AEs by maximum toxicity grade. AEs leading to dose interruption of study treatment were reported in 2 patients (bronchitis and pyrexia), of which pyrexia was considered to be related to the study treatment. No AEs leading to permanent discontinuation or dose reduction of study treatment were observed. Death due to progression of disease was reported in 1 patient. No fatal SAEs were observed. SAEs were reported in 2 patients who received study treatment, including grade 1 bronchitis (in the dabrafenib 100 mg cohort) and grade 1 myocardial ischaemia (in the dabrafenib 150 mg cohort); of these, myocardial ischaemia, which was an asymptomatic electrocardiographic change was considered to be related to the study treatment.
Pyrexia, new primary melanomas, squamous cell carcinoma or keratoacanthomas, treatment-emergent malignancies (excluding cutaneous squamous cell carcinoma (cSCC), basal cell carcinoma (BCC), and new primary melanomas), renal failure, uveitis, pancreatitis, hypersensitivity, and hyperglycemia were evaluated as AEs of special interest; of which only pyrexia, renal failure, and hypersensitivity were reported in the current study. All events of pyrexia were considered as related to the study treatment and were reported in 6 patients (1 each in the 75 mg and 100 mg cohorts and 4 patients in the 150 mg cohort). Grade 2 pyrexia occurred in 2 patients from the 150 mg cohort, and led to dose interruption in one of these 2 patients. Renal failure, which included grade 2 proteinuria and grade 1 increase in serum creatinine, was reported in 1 patient each from the 75 mg and 100 mg cohorts; both patients recovered at 15 days after event onset. Hypersensitivity was reported by 2 patients in the 100 mg cohort, of which Grade 2 contact dermatitis was not considered related to the study drug, while grade 2 urticaria was considered to be related. The time to recovery from contact dermatitis was 17 days and that for grade 2 urticaria was 59 days post occurrence.
Worsening of clinical laboratory parameters from baseline to any grade was reported in 4 out of 12 patients. Most changes were grade 1 or 2 clinical laboratory abnormalities and 1 patient reported decreased lymphocyte counts (grade 3 laboratory abnormality) at the end of the study treatment, which was reported as an AE. QT interval prolongation from a baseline of 433 msec to 457 msec was a clinically significant abnormal ECG finding reported in 1 patient by the investigator. Increased systolic and diastolic blood pressure was seen in 10 (83%) and 6 (50%) patients, respectively, and was considered as a grade 1 or 2 change.
Pharmacokinetics
Table 5 lists the PK parameters observed after single and multiple dosing with dabrafenib. Following administration of a single oral dose, there was a rapid increase in plasma dabrafenib levels, reaching a median Tmax at 3.98 h, 1.00 h, and 2.46 h, respectively, with 75 mg, 100 mg, and 150 mg dabrafenib. Following single dosing, the plasma AUC0–12 and Cmax of dabrafenib increased with dose up to 100 mg, although the exposures were similar between 100 mg and 150 mg. In the analysis using the power model, the point estimates (90% CI) of the slope of Cmax and AUC0-∞ for dabrafenib were 0.498 (−0.375 to 1.371) ng/mL and 0.763 (−0.029 to 1.555) hr.·ng/mL, respectively. Limited number of evaluable subjects and great variability led to difficulty with the evaluation for linearity.
Following multiple dosing, the AUC0–12 of plasma dabrafenib were 38%, 47% or 36% lower than those at a single dose of 75 mg, 100 mg, or 150 mg, respectively. Trough concentrations of dabrafenib 150 mg and its metabolites were considered to reach the steady state by Week 3 (Table 6), although variations were found after Week 6.
Discussion
The primary purpose of the current phase 1 study was to assess the safety and tolerability of dabrafenib in 12 Japanese patients with BRAF V600 mutation-positive solid tumors. Our study demonstrated a manageable safety profile with dabrafenib. The AEs reported in this Japanese study were similar to those reported in earlier clinical studies worldwide [22,23,24, 26]. Most AEs reported in this study were of grade 1 or 2, similar to those reported in other studies. Alopecia, pyrexia, arthralgia, leukopenia, hyperkeratosis, and nausea were AEs reported in at least 4 out of 12 patients. AEs requiring a temporary suspension of the study drug occurred in 2 patients, 1 of whom experienced drug-related pyrexia and the other experienced bronchitis not related to the study drug; both the events resolved and administration of the dabrafenib was resumed at the dose before suspension. However, it should be noted that in the absence of complications, temporary dose interruptions and symptomatic treatment have been shown to manage pyrexia and allow reintroduction of therapy in patients [27]. Pyrexia has been reported as a common AE with dabrafenib in the range of 16–26% when given as monotherapy [22,23,24] or considerable higher (51%) with combination therapy [28].
While all patients reported an AE, no patient discontinued the study because of an AE and most AEs were manageable with symptomatic treatment and did not require a dose change. Previous studies with dabrafenib [22, 26] reported a need for change in dose due to AEs such as pyrexia, fatigue, and neutropenia. Selective BRAF inhibitor therapy has been shown to be associated with development of cutaneous manifestations such as keratoacanthoma like squamous cell carcinoma, warty dyskeratomas, verrucous keratosis, acantholytic dyskeratosis. [29]. Skin reactions are commonly seen as toxic effects with dabrafenib, which necessitates frequent dermatologic examination. In the current study, class effects of BRAF inhibitors, such as squamous cell carcinoma of skin and keratoacanthoma, were not observed which have been commonly reported earlier.
The study demonstrated anti-tumor activity of dabrafenib, and response to treatment was observed in 58% of all patients. Of 11 patients with malignant melanoma, 55% showed response. An earlier phase I trial (BREAK-1) including 156 patients with metastatic melanoma reported 69% PR or CR and 50% confirmed response [22]. Response with dabrafenib monotherapy was also reported in three phase II and III studies (BREAK-2 [30], BREAK-3 [23], and BREAK-MB [24]) which included patients with BRAF V600 mutation-positive melanoma. Best overall response included PR in 3 patients in the 75 mg cohort and in 2 patients each in the 100 and 150 mg cohorts and SD in 1 patient each from the 100 and 150 mg cohorts. A dose escalation decision was made after 3 patients were enrolled in each dose cohort until a dose of 150 mg BID, the recommended/approved dose as per previous monotherapy studies, was reached. Although no protocol-defined DLTs were reported and the maximum tolerated dose (MTD) was not reached, the dose was not escalated beyond 150 mg BID based on the results from the dabrafenib 150 mg BID cohort considering that study treatment exposure was not expected to increase in a higher dose cohort as PK analyses revealed no differences in exposure in the 100 mg and 150 mg cohorts. Further, tumor response was seen in all the dose cohorts.
Dabrafenib was rapidly absorbed after single and multiple oral dosing. Increase in plasma AUC0–12 and Cmax was seen with both single and multiple doses and was associated with a dose increase from 75 mg to 100 mg BID and was comparable between the 100 mg and 150 mg cohorts. Dose proportionality could not be determined by the power model analysis due to the small sample size and large variation observed in data. There were no significant differences between the PK profile observed in this study and trials reported earlier. Since dabrafenib shows auto-induction, plasma AUC0–12 at multiple doses was approximately 40% lower than that at a single dose.
BRAF inhibitors such as vemurafenib and dabrafenib have demonstrated impressive results in phase I, II, and III trials and are considered as the standard of care in the treatment of BRAF-mutation positive metastatic melanoma [31]. While dabrafenib and vemurafenib are similar in several aspects such as both being selective type 1 BRAF inhibitors, in clinical efficacy, etc.; they differ in terms of RAF kinase inhibition, toxicities and activity in non-V600E BRAF melanoma and in brain metastasis [32]. Both dabrafenib and vemurafenib have similar inhibitory potency for the RAF proto-oncogene serine/threonine-protein kinase (c-RAF) as well as BRAF V600E [33, 34]. However dabrafenib has been demonstrated to be a more selective inhibitor of BRAF V600E than vemurafenib, based on the potency of c-RAF and BRAFV600E inhibition compared to wild type BRAF (BRAFwt) inhibition (IC50 ratio of 0.4 and 0.05 with dabrafenib [34] compared to 0.5 and 0.3 with vemurafenib [33]). Further, dabrafenib has also shown similar potency for inhibition of BRAF V600E and BRAF V600K [22]. As a result of low incidence, there is a lack of sufficient medical knowledge about the disease, and most patients are diagnosed at the stage of advanced disease [35], which is challenging to conventional treatment modalities, thereby increasing the importance of novel therapies such as BRAF inhibitors. The current study was limited by sample size, as is expected in a first Japanese phase I study, however, 11 out of 12 Japanese patients in the study had melanoma. Dabrafenib 150 mg BID, which is the recommended dose worldwide, was proven to be well tolerated by Japanese patients, with no marked differences in the safety and PK profile compared with previous clinical studies.
In summary, 12 Japanese patients with BRAF mutation positive solid cancers participated in this study and were treated with 75 mg BID, 100 mg BID, or 150 mg BID of dabrafenib. The study concluded similar safety, tolerability, efficacy, and PK to Caucasian patients who were treated with 150 mg BID of dabrafenib.
References
Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JW, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA (2002) Mutations of the BRAF gene in human cancer. Nature 417(6892):949–954. doi:10.1038/nature00766
Puxeddu E, Filetti S (2014) BRAF mutation assessment in papillary thyroid cancer: are we ready to use it in clinical practice? Endocrine 45(3):341–343. doi:10.1007/s12020-013-0139-0
Clancy C, Burke JP, Kalady MF, Coffey JC (2013) BRAF mutation is associated with distinct clinicopathological characteristics in colorectal cancer: a systematic review and meta-analysis. Color Dis 15(12):e711–e718. doi:10.1111/codi.12427
Tiacci E, Trifonov V, Schiavoni G, Holmes A, Kern W, Martelli MP, Pucciarini A, Bigerna B, Pacini R, Wells VA, Sportoletti P, Pettirossi V, Mannucci R, Elliott O, Liso A, Ambrosetti A, Pulsoni A, Forconi F, Trentin L, Semenzato G, Inghirami G, Capponi M, Di Raimondo F, Patti C, Arcaini L, Musto P, Pileri S, Haferlach C, Schnittger S, Pizzolo G, Foa R, Farinelli L, Haferlach T, Pasqualucci L, Rabadan R, Falini B (2011) BRAF mutations in hairy-cell leukemia. N Engl J Med 364(24):2305–2315. doi:10.1056/NEJMoa1014209
Brastianos PK, Taylor-Weiner A, Manley PE, Jones RT, Dias-Santagata D, Thorner AR, Lawrence MS, Rodriguez FJ, Bernardo LA, Schubert L, Sunkavalli A, Shillingford N, Calicchio ML, Lidov HG, Taha H, Martinez-Lage M, Santi M, Storm PB, Lee JY, Palmer JN, Adappa ND, Scott RM, Dunn IF, Laws ER Jr, Stewart C, Ligon KL, Hoang MP, Van Hummelen P, Hahn WC, Louis DN, Resnick AC, Kieran MW, Getz G, Santagata S (2014) Exome sequencing identifies BRAF mutations in papillary craniopharyngiomas. Nat Genet 46(2):161–165. doi:10.1038/ng.2868
Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, Cho KH, Aiba S, Brocker EB, LeBoit PE, Pinkel D, Bastian BC (2005) Distinct sets of genetic alterations in melanoma. N Engl J Med 353(20):2135–2147. doi:10.1056/NEJMoa050092
Bhatia P, Friedlander P, Zakaria EA, Kandil E (2015) Impact of BRAF mutation status in the prognosis of cutaneous melanoma: an area of ongoing research. Ann Transl Med 3(2):24. doi:10.3978/j.issn.2305-5839.2014.12.05
Maurer G, Tarkowski B, Baccarini M (2011) Raf kinases in cancer-roles and therapeutic opportunities. Oncogene 30(32):3477–3488. doi:10.1038/onc.2011.160
Ascierto PA, Kirkwood JM, Grob JJ, Simeone E, Grimaldi AM, Maio M, Palmieri G, Testori A, Marincola FM, Mozzillo N (2012) The role of BRAF V600 mutation in melanoma. J Transl Med 10:85. doi:10.1186/1479-5876-10-85
Sakaizawa K, Ashida A, Uchiyama A, Ito T, Fujisawa Y, Ogata D, Matsushita S, Fujii K, Fukushima S, Shibayama Y, Hatta N, Takenouchi T, Uehara J, Okuyama R, Yamazaki N, Uhara H (2015) Clinical characteristics associated with BRAF, NRAS and KIT mutations in Japanese melanoma patients. J Dermatol Sci 80(1):33–37. doi:10.1016/j.jdermsci.2015.07.012
Fukushima T, Suzuki S, Mashiko M, Ohtake T, Endo Y, Takebayashi Y, Sekikawa K, Hagiwara K, Takenoshita S (2003) BRAF mutations in papillary carcinomas of the thyroid. Oncogene 22(41):6455–6457. doi:10.1038/sj.onc.1206739
Ito Y, Yoshida H, Maruo R, Morita S, Takano T, Hirokawa M, Yabuta T, Fukushima M, Inoue H, Tomoda C, Kihara M, Uruno T, Higashiyama T, Takamura Y, Miya A, Kobayashi K, Matsuzuka F, Miyauchi A (2009) BRAF mutation in papillary thyroid carcinoma in a Japanese population: its lack of correlation with high-risk clinicopathological features and disease-free survival of patients. Endocr J 56(1):89–97
Namba H, Nakashima M, Hayashi T, Hayashida N, Maeda S, Rogounovitch TI, Ohtsuru A, Saenko VA, Kanematsu T, Yamashita S (2003) Clinical implication of hot spot BRAF mutation, V599E, in papillary thyroid cancers. J Clin Endocrinol Metab 88(9):4393–4397. doi:10.1210/jc.2003-030305
Konishi K, Takimoto M, Kaneko K, Makino R, Hirayama Y, Nozawa H, Kurahashi T, Kumekawa Y, Yamamoto T, Ito H, Yoshikawa N, Kusano M, Nakayama K, Rembacken BJ, Ota H, Imawari M (2006) BRAF mutations and phosphorylation status of mitogen-activated protein kinases in the development of flat and depressed-type colorectal neoplasias. Br J Cancer 94(2):311–317. doi:10.1038/sj.bjc.6602911
Yokota T, Ura T, Shibata N, Takahari D, Shitara K, Nomura M, Kondo C, Mizota A, Utsunomiya S, Muro K, Yatabe Y (2011) BRAF mutation is a powerful prognostic factor in advanced and recurrent colorectal cancer. Br J Cancer 104(5):856–862. doi:10.1038/bjc.2011.19
Nakayama N, Nakayama K, Yeasmin S, Ishibashi M, Katagiri A, Iida K, Fukumoto M, Miyazaki K (2008) KRAS or BRAF mutation status is a useful predictor of sensitivity to MEK inhibition in ovarian cancer. Br J Cancer 99(12):2020–2028. doi:10.1038/sj.bjc.6604783
Eisen T, Ahmad T, Flaherty KT, Gore M, Kaye S, Marais R, Gibbens I, Hackett S, James M, Schuchter LM, Nathanson KL, Xia C, Simantov R, Schwartz B, Poulin-Costello M, O'Dwyer PJ, Ratain MJ (2006) Sorafenib in advanced melanoma: a phase II randomised discontinuation trial analysis. Br J Cancer 95(5):581–586. doi:10.1038/sj.bjc.6603291
Ott PA, Hamilton A, Min C, Safarzadeh-Amiri S, Goldberg L, Yoon J, Yee H, Buckley M, Christos PJ, Wright JJ, Polsky D, Osman I, Liebes L, Pavlick AC (2010) A phase II trial of sorafenib in metastatic melanoma with tissue correlates. PLoS One 5(12):e15588. doi:10.1371/journal.pone.0015588
Su Y, Vilgelm AE, Kelley MC, Hawkins OE, Liu Y, Boyd KL, Kantrow S, Splittgerber RC, Short SP, Sobolik T, Zaja-Milatovic S, Dahlman KB, Amiri KI, Jiang A, Lu P, Shyr Y, Stuart DD, Levy S, Sosman JA, Richmond A (2012) RAF265 inhibits the growth of advanced human melanoma tumors. Clin Cancer Res 18(8):2184–2198. doi:10.1158/1078-0432.ccr-11-1122
Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, Jouary T, Schadendorf D, Ribas A, O'Day SJ, Sosman JA, Kirkwood JM, Eggermont AM, Dreno B, Nolop K, Li J, Nelson B, Hou J, Lee RJ, Flaherty KT, McArthur GA (2011) Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364(26):2507–2516. doi:10.1056/NEJMoa1103782
Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, McArthur GA, Hutson TE, Moschos SJ, Flaherty KT, Hersey P, Kefford R, Lawrence D, Puzanov I, Lewis KD, Amaravadi RK, Chmielowski B, Lawrence HJ, Shyr Y, Ye F, Li J, Nolop KB, Lee RJ, Joe AK, Ribas A (2012) Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med 366(8):707–714. doi:10.1056/NEJMoa1112302
Falchook GS, Long GV, Kurzrock R, Kim KB, Arkenau TH, Brown MP, Hamid O, Infante JR, Millward M, Pavlick AC, O'Day SJ, Blackman SC, Curtis CM, Lebowitz P, Ma B, Ouellet D, Kefford RF (2012) Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet 379(9829):1893–1901. doi:10.1016/s0140-6736(12)60398-5
Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, Rutkowski P, Blank CU, Miller WH Jr, Kaempgen E, Martin-Algarra S, Karaszewska B, Mauch C, Chiarion-Sileni V, Martin AM, Swann S, Haney P, Mirakhur B, Guckert ME, Goodman V, Chapman PB (2012) Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 380(9839):358–365. doi:10.1016/s0140-6736(12)60868-x
Long GV, Trefzer U, Davies MA, Kefford RF, Ascierto PA, Chapman PB, Puzanov I, Hauschild A, Robert C, Algazi A, Mortier L, Tawbi H, Wilhelm T, Zimmer L, Switzky J, Swann S, Martin AM, Guckert M, Goodman V, Streit M, Kirkwood JM, Schadendorf D (2012) Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicentre, open-label, phase 2 trial. Lancet Oncol 13(11):1087–1095. doi:10.1016/s1470-2045(12)70431-x
King AJ, Arnone MR, Bleam MR, Moss KG, Yang J, Fedorowicz KE, Smitheman KN, Erhardt JA, Hughes-Earle A, Kane-Carson LS, Sinnamon RH, Qi H, Rheault TR, Uehling DE, Laquerre SG (2013) Dabrafenib; preclinical characterization, increased efficacy when combined with trametinib, while BRAF/MEK tool combination reduced skin lesions. PLoS One 8(7):e67583. doi:10.1371/journal.pone.0067583
Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N, Kudchadkar R, Burris HA 3rd, Falchook G, Algazi A, Lewis K, Long GV, Puzanov I, Lebowitz P, Singh A, Little S, Sun P, Allred A, Ouellet D, Kim KB, Patel K, Weber J (2012) Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med 367(18):1694–1703. doi:10.1056/NEJMoa1210093
Welsh SJ, Corrie PG (2015) Management of BRAF and MEK inhibitor toxicities in patients with metastatic melanoma. Ther Adv Med Oncol 7(2):122–136. doi:10.1177/1758834014566428
Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, Garbe C, Jouary T, Hauschild A, Grob JJ, Chiarion Sileni V, Lebbe C, Mandala M, Millward M, Arance A, Bondarenko I, Haanen JB, Hansson J, Utikal J, Ferraresi V, Kovalenko N, Mohr P, Probachai V, Schadendorf D, Nathan P, Robert C, Ribas A, DeMarini DJ, Irani JG, Casey M, Ouellet D, Martin AM, Le N, Patel K, Flaherty K (2014) Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med 371(20):1877–1888. doi:10.1056/NEJMoa1406037
Chu EY, Wanat KA, Miller CJ, Amaravadi RK, Fecher LA, Brose MS, McGettigan S, Giles LR, Schuchter LM, Seykora JT, Rosenbach M (2012) Diverse cutaneous side effects associated with BRAF inhibitor therapy: a clinicopathologic study. J Am Acad Dermatol 67(6):1265–1272. doi:10.1016/j.jaad.2012.04.008
Ascierto PA, Minor D, Ribas A, Lebbe C, O'Hagan A, Arya N, Guckert M, Schadendorf D, Kefford RF, Grob JJ, Hamid O, Amaravadi R, Simeone E, Wilhelm T, Kim KB, Long GV, Martin AM, Mazumdar J, Goodman VL, Trefzer U (2013) Phase II trial (BREAK-2) of the BRAF inhibitor dabrafenib (GSK2118436) in patients with metastatic melanoma. J Clin Oncol 31(26):3205–3211. doi:10.1200/jco.2013.49.8691
Scholtens A, Geukes Foppen MH, Blank CU, van Thienen JV, van Tinteren H, Haanen JB (2015) Vemurafenib for BRAF V600 mutated advanced melanoma: results of treatment beyond progression. Eur J Cancer 51(5):642–652. doi:10.1016/j.ejca.2015.01.009
Menzies AM, Long GV, Murali R (2012) Dabrafenib and its potential for the treatment of metastatic melanoma. Drug Des Devel Ther 6:391–405. doi:10.2147/dddt.s38998
Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, Spevak W, Zhang C, Zhang Y, Habets G, Burton EA, Wong B, Tsang G, West BL, Powell B, Shellooe R, Marimuthu A, Nguyen H, Zhang KY, Artis DR, Schlessinger J, Su F, Higgins B, Iyer R, D'Andrea K, Koehler A, Stumm M, Lin PS, Lee RJ, Grippo J, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, Chapman PB, Flaherty KT, Xu X, Nathanson KL, Nolop K (2010) Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 467(7315):596–599. doi:10.1038/nature09454
Kefford R, Arkenau H, Brown MP, et al (2010) Phase I/II study of GSK2118436, a selective inhibitor of oncogenic mutant BRAF kinase, in patients with metastatic melanoma and other solid tumors. J Clin Oncol 28 (S611):abstract 8503
Yoichi Moroi (2013) Patient Surveillance After Cancer Treatment,vol 1. doi:10.1007/978-1-60327-969-7_52
Acknowledgements
The study was funded by GlaxoSmithKline. Dabrafenib is an asset of Novartis AG as of March 2, 2015. We thank all the investigators and patients who participated in the study, their families as well as staff at the study centers. We also thank Dr. Krishna Swetha Gummuluri, Novartis Healthcare Pvt. Ltd., India, for medical writing support.
Funding
GlaxoSmithkline
Author information
Authors and Affiliations
Contributions
All of the authors had full access to data in the study, discussed the results, critically reviewed the draft manuscript, and approved the final manuscript for submission
Corresponding author
Ethics declarations
Conflict of interest
Y Fujiwara reports grants from AstraZeneca, Chugai, Daiichi-Sankyo, Eisai, Eli Lilly, GlaxoSmithKline, Merck Serono, MSD and Novartis; and honoraria and personal fees from Bristol-Myers Squibb and ONO. N Yamazaki reports personal fees from ONO, Takeda, Bristol-Myers Squibb, Chugai, Boehringer Ingelheim and research funding from ONO, Bristol-Myers Squibb and Novartis Pharma K.K. Y Kiyohara reports personal fees from ONO, Chugai, Bristol-Myers Squibb and grants from Chugai and MSD. N Yamamoto reports grants from Chugai,Taiho, Eisai, Eli Lilly, Quintiles, Astellas, BMS, Novartis, Daiichi-Sankyo, Pfizer,Boehringer Ingelheim, Kyowa-Hakko Kirin, Bayer, ONO and Takeda and other support from Chugai, Astrazeneca, Eli Lilly and BMS. H Nokihara reports grants from Merck Serono, Pfizer, Novartis, Daiichi Sankyo, GlaxoSmithKline and Quintiles and personal fees from Taiho Pharmaceuticals, Eisai, Chugai, Eli Lilly, AstraZeneca, Boehringer-Ingelheim,ONO, Sanofi and Bristol-Myers Squibb. K Namikawa reports personal fees from ONO, Bristol-Myers Squibb, Merck Sharp and Dohme, Toray Industries, Chugai and Novartis Pharmaceuticals. T Tamura reports personal fees from Chugai, Taiho, Eisai, Yakult, Eli Lilly, Boehringer-Ingelheim, Bristol-Myers Squibb, ONO and Kyowa Kirin. A Mukaiyama and F Zhang are employees of Novartis Pharma K.K., Japan. S Yoshikawa, and A Tsutsumida have nothing to disclose.
Ethical approval
All procedures performed in the study were conducted in accordance with the International Conference on Harmonization (ICH) Guidelines for Good Clinical Practice (GCP) and the Declaration of Helsinki. All protocols and amendments were approved by the independent ethics committee or institutional review board for each study center. All patients provided written informed consent before participating in any study procedures.
Rights and permissions
About this article

Cite this article
Fujiwara, Y., Yamazaki, N., Kiyohara, Y. et al. Safety, tolerability, and pharmacokinetic profile of dabrafenib in Japanese patients with BRAF V600 mutation-positive solid tumors: a phase 1 study. Invest New Drugs 36, 259–268 (2018). https://doi.org/10.1007/s10637-017-0502-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-017-0502-8