
Summary
Background
Acute lymphoblastic leukemia (ALL) is a potentially fatal disease that involves clonal expansion of early lymphoid progenitor cells. Much of drug development for ALL treatment involves targeting antigens of the clonal cell surface. Blinatumomab belongs to an emerging class of anti-cancer therapeutics referred to as bispecific T-cell engaging antibodies. The Food and Drug Administration approved its use in relapsed or refractory adult Philadelphia chromosome-negative B-cell precursor ALL in December of 2014.
Mechanism of action and pharmacodynamics
Blinatumomab contains both an anti-CD3 and anti-CD19 arm, allowing for the juxtaposition of CD3+ T-cells to malignant CD19+ B-cells, thereby resulting in granzyme- and perforin-mediated B-cell apoptosis.
Preclinical pharmacology
Preclinical studies suggest that blinatumomab’s efficacy is related to the effector-to-target ratio and to the difference between its affinity for CD19 and CD3.
Pharmacokinetics and metabolism
Preclinical and early phase clinical studies have allowed for the characterization of the pharmacokinetics of blinatumomab, including the determination of its short half-life. The metabolic pathway has not been fully characterized but is thought to be similar to that of other antibodies.
Clinical studies
Phase I and II studies led to the identification of an ideal stepwise dose, involving long-term continuous intravenous infusion (CIVI), to optimize its efficacy and reduce the risk of certain toxicities. A high remission rate and duration were noted among a relapsed/refractory population of patients.
Safety
The results of clinical trials have identified cytokine release syndrome and neurotoxicity, among others, as serious drug-related toxicities, leading to the institution of a Risk Evaluation and Mitigation Strategy.
Discussion and conclusions
Blinatumomab represents a significant addition to the treatment options for ALL, but it is not without its limitations, of which are its short-half life, necessitating long-term CIVI, and the eventual emergence of CD19-negative clones. Continual development of the agent involves assessing its role in the frontline setting and in combination with chemotherapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Acute lymphoblastic leukemia (ALL) is a heterogeneous group of diseases involving clonal expansion of early lymphoid progenitor cells, invariably leading to death unless immediately treated. In the United States, the incidence of ALL is approximately 17 cases per million individuals (6020 new cases per year), 40 % of whom are over 20 years in age [1, 2]. The 2008 revision of the World Health Organization Classification of Tumors of Hematopoietic and Lymphoid Tissues classifies ALL as either B- or T-lymphoblastic leukemia, with further classification of B-ALL into distinct entities defined by specific chromosomal abnormalities [3]. A more simplistic and clinically practical classification for precursor ALL, however, is to broadly categorize B-ALL into Philadelphia chromosome-negative (Ph-neg) ALL, accounting for approximately 75 % of adult ALL cases, and Philadelphia chromosome-positive (Ph-pos) ALL which accounts for approximately 25 % of adult ALL cases [4]. Frontline induction therapy for ALL (for the remainder of the article, “ALL” will refer to precursor ALL, as opposed to mature ALL) often involves multiagent cytotoxic chemotherapy regimens, consisting of an anthracycline, a vinca alkaloid, cyclophosphamide, and a corticosteroid. Etoposide, methotrexate (MTX), cytarabine (Ara-C), and asparaginase are also commonly incorporated into frontline therapeutic therapy, as is central nervous system (CNS) prophylaxis (or therapy) in the form of intrathecal or intraventricular MTX and/or Ara-C [5]. The addition of rituximab, a chimeric anti-CD20 monoclonal antibody (mAb) used to treat various B-cell neoplasms, to chemotherapy has been shown to improve outcomes in CD20+ precursor B-cell ALL patients younger than age 55–60 years [6, 7]. In Ph-pos ALL, it has become standard practice to combine a BCR-ABL tyrosine kinase inhibitor (TKI) with induction chemotherapy, supported by the results of studies that suggest improved response rates and survival as compared to historical controls [8].
After attainment of a morphologic complete remission (CR), which occurs in over 80 % of patients, consolidation/intensification followed by maintenance therapy is common practice, as outlined by the National Comprehensive Cancer Network [5], unless allogeneic hematopoietic stem cell transplant (alloSCT) is an option for appropriate high-risk patients. Despite the relatively high CR rates associated with frontline therapy, the 5-year survival among adolescents and adults collectively is approximately 40 % [9], as opposed to 94 % among the pediatric population [10]. This outcome discrepancy between adults and children may be attributable to differences in disease biology, treatment approach, and tolerance to therapy [11]. The persistence of minimal residual disease (MRD), possibly represented by quiescent leukemia initiating cells (LICs) resistant to conventional cytotoxic therapy [12], may explain the high rate of relapse among adults, having prompted clinical investigators to incorporate MRD assessment into protocols during various time points after induction therapy [13]. For relapsed or refractory (R/R) ALL, salvage therapy may result in CR rates of less than 20 % with median duration of response of 7 months, median overall survival (OS) of 3 months, and a 5-year OS of 7 % [14, 15]. Currently available salvage options include combination cytotoxic chemotherapy (e.g., FLAG), clofarabine, an alternative TKI for Ph-pos ALL, nelarabine for T-ALL, vincristine sulfate liposome injection for Ph-neg ALL, and now blinatumomab for R/R, Ph-neg, precursor B-ALL in adults. Efforts to improve outcomes for adult ALL patients involve the use of pediatric-inspired regimens (often asparaginase-based) [16], liposomal forms of standard drugs in addition to vincristine [17], novel purine nucleoside analogs [18], and most promisingly, the development of targeted agents.
Development of novel, targeted agents currently primarily focuses on treatment of R/R disease, but there are efforts to study these agents as consolidative/maintenance therapies in an effort to control or eradicate MRD after attainment of morphologic CR (from standard induction therapy) and to incorporate these agents into frontline, multiagent induction regimens. Under clinical investigation, as single agents or as components of combination regimens, are the small molecule inhibitors of various intracellular targets, such as Notch, aurora kinase, histone deacetylase, and mammalian target of rapamycin (mTOR). However, the investigational targeted agents that are furthest in development are mAb-based drugs that target leukemic blast cell surface antigens, particularly CD19 and CD22 [19, 20]. Targeting these commonly observed cell surface antigens may allow for an otherwise genetically heterogeneous population to benefit from the same drug, particularly considering that CD19 is expressed in more than 90 % of cases of B-ALL and CD22 in more than 80 % [20]. Studies of the CD22-targeting agents epratuzumab, a humanized naked mAb, and the antibody-drug conjugate (ADC) inotuzumab ozogamicin have demonstrated activity in adult ALL [20, 21]. In addition to blinatumomab, the ADC SAR3419 and chimeric antigen receptor (CAR) T-cells that are engineered to target CD19 are among the growing list of anti-CD19 agents being developed for ALL treatment [22, 23].
CD19 is a 95-kDa transmembrane B-cell-specific coreceptor, belonging to the immunoglobulin superfamily, that enhances signaling through the B-cell receptor (BCR), thereby regulating B-cell survival and differentiation [20, 24]. It was identified (referred to as B4) in the early 1980s as a B cell-specific antigen appearing early in B-cell development, increasing with mitogen-stimulation of B-cells, and then becoming lost at the terminal stage of differentiation [25]. It may have dual roles in B-cell activation, serving as an adaptor protein to recruit cytoplasmic signaling proteins to the membrane and regulating bone marrow development by altering BCR signals [26]. CD19 lowers the threshold for antigen receptor stimulation of B-cells, contributing to the efficiency of the immune system in which unstimulated B-cells need to respond to low levels of antigen for the efficient elimination of infections [27]. Its function is highlighted by the recognition that individuals with germline homozygous CD19 gene mutations may have normal numbers of mature B-cells albeit with a defective response to antigenic stimulation, with associated hypogammaglobulinemia, and susceptibility to infection [28]. CD19 also represents the most commonly expressed antigen in pre-B-ALL, as noted from immunophenotyping studies in a series of 451 cases of B-ALL in which all cases were positive for CD19 [29]. CD19 is the most densely expressed of the B-ALL cell surface antigens. Because it is ubiquitously present on B-cells throughout early and late stages of differentiation but absent among hematopoietic stem cells and plasma cells, CD19 is a highly attractive target for drug development in B-ALL [24, 26].
Proof of principle of effective therapeutic targeting of CD19 in B-ALL has been shown in many preclinical studies dating back several decades. Even as far back as the late 1980s, it was recognized by scientists that CD19 provides an opportunity to deliver mAb-bound toxins (i.e., immunoconjugates) into leukemia cells, upon learning that CD19 receptor-ligand complexes become internalized and enter endosomes [30]. In severe combined immunodeficiency (SCID) mice engrafted with a human ALL cell line, administration of the immunotoxin B43 (anti-CD19)-pokeweed antiviral protein decreased morbidity and improved survival, whereas an anti-CD4 immunotoxin did not in these mice [31]. Another study also involving a SCID mouse model engrafted with human ALL cells demonstrated activity of a single injection of radiolabeled anti-CD19 mAb with delayed suppression on the level of circulating leukemic cells and improvement in median survival [32]. These studies and multiple others have paved the way for the clinical development of anti-CD19 therapy for the treatment of B-ALL.
Blinatumomab, derived from murine B-cell antibodies and belonging to an emerging class of therapies referred to as bispecific T-cell engaging (BiTE®) antibodies, is a 55-kDa single-chain antibody that contains both an anti-CD3 and anti-CD19 arm that are joined by a non-immunogenic linker (Fig. 1). The use of recombinant DNA technology using the cDNA sequences that encode for the variable domains and linker sequences allows for its novel design [33]. Its single-chain structure allows for rotational flexibility with binding two different cell types in close proximity and for the ability to produce large amounts of this agent in a pure and stable form [34]. The concept of bispecific antibody constructs are not entirely new, however. In 1985, Staerz et al. described a heteroconjugate of two antibodies, one of which targeted cytotoxic T-cells and the other of which targeted a cell surface antigen of cancer cells, resulting in tumor cell lysis [35]. In the 1990s, Mack et al. reported their construction of and experience with a bispecific single-chain antibody derivative, involving two different antibodies’ Fv fragments joined via a linker molecule [36]. This construct directed cytotoxic T-cells to 17-1A-positive tumor cells, resulting in high degrees of cytotoxicity with nanomolar concentrations of the agent. Currently in development, in addition to blinatumomab, are a number of BiTE antibodies that redirect CD3+ T-cells to cell surface antigens of cells of various tumor histologies [37].

Blinatumomab is a 55-kDA single-chain, bispecific T-cell engaging antibody that contains Fv fragements from both an anti-CD3 and anti-CD19 arm joined by a non-immunogenic linker, providing rotational flexibility that allows for the juxtaposition of cytotoxic CD3+ T-cells to malignant CD19+ B-cells, thereby resulting in granzyme- and perforin-mediated B-cell apoptosis
Mechanism of action and pharmacodynamics
Blinatumomab, also referred to as bscCD19xCD3, is composed of two single chain distinct parental murine mAbs: one which recognizes the B cell antigen, CD19 and another which binds the T-cell receptor associated complex, CD3 [38]. They are linked by a glycine/serine amino acid complex, which allows for a high degree of flexibility needed for simultaneous binding of two cells [33]. Blinatumomab, by engaging these two molecules, facilitates the formation of a transient cytolytic synapse between T-cells and malignant B-cells, leading to activation of serial target cell lysis that resembles natural T-cell mediated killing. Engaged T-cells release perforins and granzymes from their secretory vehicles into target cells to prompt nuclear fragmentation and membrane blebbing (i.e., programmed cell death). T-cell activation induces transient release of cytokines that leads to T-cell activity alteration from scanning to killing mode and T-cell proliferation [39]. Notably, the activity of blinatumomab is independent of antigen presentation by class I MHC and TCR recognition. Therefore, blinatumomab can circumvent a variety of tumor-mediated immune escape mechanisms, such as impairment of antigen presentation machinery and activation of negative costimulatory signals in the tumor microenvironment [39, 40].
Studies have shown that shortly after continuous intravenous infusion (CIVI) of blinatumomab at doses above 5 μg/m2/day, the circulating malignant B-cell count was reduced significantly to undetectable levels [38, 41]. The B-cell count did not rebound when the CIVI was discontinued for 1 week. During the first few days of treatment, the redistribution of T-cells into the tissue resulted in a transient decline of their count with recovery noted within 2 weeks. This rebound is possibly due to increase proliferation of T-cells initiated by the increase in cytokine [38]. The increase of cytokine (IL-10, IL-6, IFN-γ, IL-2, and TNF-α) concentration is noted to be the highest in the first 48 h of continuous intravenous infusion and then returns to normal in 24 to 48 h [38].
Preclinical pharmacology
Multiple in vitro and in vivo studies have investigated blinatumomab’s ability to effect T-cell-mediated lysis of malignant B-cells [38, 39]. The potency of blinatumomab was studied with a cell culture cytotoxicity assay in which peripheral blood mononuclear (PBMC) T-cells isolated from healthy human donors were subjected to human CD19+ B-cell lymphoma cell lines. Half-maximum target lysis (EC50) varied based on the effector-to-target (E:T) ratio, with most cell lysis taking place at an E:T of 10:1 [38]. In a later study that explored the efficacy of blinatumomab at an E:T ≤ 1:1, the EC50 ranged from 20 to 200 pg/ml with E:T ratios 1:1 and 1:10, respectively, indicating serial cell killing by T cells in the presence of blinatumomab [39]. Three key factors seemed to determine the level of the therapeutic effect: T cell activity, E:T ratio, and the time needed to create a cytolytic synapse. Twenty four hours was required for near maximal cell lysis with an E:T ratio of 1:5 at nanomolar concentrations of blinatumomab [39]. This high potency of blinatumomab was due to its binding affinity with CD3 and CD19. It has a much weaker disassociation constant to CD19 (Kd = 1.49 × 10−9 M) as compared to CD3 (Kd = 2.6 × 10−7 M), allowing the conjugated T-cell, at an E:T ratio as low as 1:5, to cycle through several B-cells with enough time for cytolytic activity. On the other hand, compared to previous BiTE molecules, blinatumomab has a lower affinity to CD3, minimizing T-cell-target clustering and allowing increased T-cell movement and targeted killing [38].
CD19-negative cell lines in the presence of T cells at an E:T ratio of 10:1 were unaffected even at high blinatumomab concentration (100 ng/ml), demonstrating the specificity of blinatumomab to target cells [38]. The effect of corticosteroids on blinatumomab’s ability to mediate T-cell cytokine release and redirect lysis was also studied in vitro. Cytokine release by blinatumomab activated T-cells was reduced by dexamethasone at 3 × 10−7 M (corresponding to an oral dexamethasone dose of 8 mg), but this did not decrease blinatumomab’s cytolytic activity [42]. The IV administration of 0.1 or 1.0 μg of blinatumomab daily for 5 consecutive days, inhibited tumor formation in non-obese diabetic/SCID mice [39]. These mice were injected subcutaneously with a mixture of human PBMCs and CD19+ human B-ALL cell lines immediately before the administration of blinatumomab [43]. In a leukemic mouse model, the administration of blinatumomab delayed tumor growth and prolonged survival [43].
Pharmacokinetics and metabolism
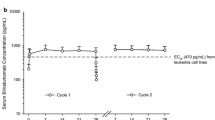
Pharmacokinetics (PKs) from preclinical and early clinical studies have shown that blinatumomab is rapidly eliminated, with a half-life of 2 h, and does not result in accumulation following multiple doses [44]. Therefore, administration of blinatumomab via CIVI has the advantage of maintaining steady drug concentration throughout the cycle. The PK parameters of blinatumomab were assessed in 116 patients enrolled in 3 clinical studies involving CIVI at a dose range of 5–90 μg/m2/day over 4–8 weeks per cycle [45]. PKs of blinatumomab demonstrated steady-state concentration (Css) that was achieved within a day and steadily maintained over 5 dosing cycles. There was also a proportional correlation between Css and dose. A dose of 5 μg/m2/day was associated with a mean Css (± SD) of 211 (±258) pg/mL, a biologically active range needed for cell lysis (EC50 was in the range of 20–200 pg/ml), and 15 μg/m2/day dosing yielded a Css of 621 (±502) pg/mL. The estimated mean clearance, mean terminal half-life, and mean volume of distribution (Vz) at the terminal phase was 2.92 (±2.83) L/hour, 2.11 (±1.42) hours, and 4.52 (±2.89) L, respectively. Also of note, in this population, with an age range of 18–80 years, weight range of 44–134 kg, and a BSA range of 1.39–2.57 m2, age, body weight, and BSA did not affect blinatumomab’s PKs [45]. BSA-based or weight-based dosing did not reduce the high inter-patient variability of systemic exposure [44]. Therefore, the fixed dose regimen used in some of the trials was justified over BSA-based dosing. Complete B-cell suppression was maintained at doses of 15 μg/m2/day, and with a mean BSA of 1.9 m2, the fixed dose used in later studies was determined to be 28 μg/day [45, 46].
The preliminary PK analyses in the pediatric population showed values similar to those of the adult studies [44, 45]. Blinatumomab was studied in pediatric patients with R/R ALL at doses ranging from 3.75 to 60 μg/m2/day, administered as CIVI for 4 weeks followed by 2 weeks rest, in a phase I/II study (MT103-205) [47]. Preliminary phase I data from PK assessments showed comparable Css values when evaluating two pediatric age groups (ages 2–6 and 7–17 y/o) at the equivalent dose level [47]. Among both age groups, mean (SD) Css was 165 (±138) pg/ml and 496 (±312) pg/ml at doses 5 μg/m2/day and 15 μg/m2/day, respectively. Mean Vz at the terminal phase, mean terminal half-life, and mean systemic clearance were 3.99 (±331) L, 2.19 (±1.53) hours, and 2.01 (±2.08) L/h, respectively. The maximum tolerated dose (MTD) was defined as 15 μg/m2/day (escalated from 5 μg/m2/day after 7 days). However, no labeling dosing recommendations were proposed for the pediatric population, given the small number of subjects in the review [47].
The metabolic pathway of blinatumomab has not yet been characterized. It is likely that blinatumomab gets degraded, like other antibodies, to small peptides and amino acids via cellular catabolic pathways [47]. There is limited renal excretion of blinatumomab. Small amounts (0.2 %) of blinatumomab have been noted in the urine of patients who received 60 μg/ m2/day dose. Formal PK studies in patients with renal impairment have not been conducted. Based on data from various trials, there is a 2-fold increase in mean clearance time in patients with moderate renal dysfunction (CrCl 30–59 ml/min) [47]. The mean (SD) clearance was 3.26 (±3.11) L/h, 2.22 (±1.76) L/h and 1.58 (±0.98) L/h for patients with CrCL ≥ 90 mL/min, 60–89 mL/min, and 30–59 mL/min, respectively. However, high inter-subject variability in clearance was noted, with a constant variance of 95.6 %, which supported recommendation for no dosage adjustment for mild to moderate renal impairment [44, 47]. A higher rate of adverse events and treatment discontinuation were noted in patients with decreased renal function, but advanced age was a confounding factor [44]. Patients with severe renal dysfunction (CrCl < 30 mL/min and on dialysis) have not been enrolled in the studies, and therefore no dose recommendations have been made for these patients.
Clinical studies
Phase I trials
Blinatumomab entered into clinical trials in 2001, at which time three first-in-human, dose escalation phase I trials (MT103 1/01-2001, −2002, and −2003) in Germany and Sweden involving a total of 21 patients with relapsed or refractory (R/R) non-Hodgkin lymphoma (NHL) or chronic lymphocytic leukemia were conducted [33]. Blinatumomab was given as short-term infusions at doses ranging from 0.75 to 13 μg/m2 as frequently as once to three times per week. Disappointingly, there were no signals of activity among these patients, and given the significant toxicities observed, notably neurologic adverse events (AEs) and cytokine release syndrome (CRS), these trials were terminated early. The experience from these trials and the knowledge of the short half life of the drug (~2 h) in humans prompted further investigation of blinatumomab as a CIVI over at least 4 weeks, made possible with the use of a small portable infusion pump [33]. This general dosing schedule has been used in all of the subsequent blinatumomab trials (Table 1). A 2004 German dose escalation phase I study involving 76 patient with relapsed and refractory NHL (primary refractory, n = 48), including indolent, mantle cell, and diffuse large B-cell lymphomas (DLBCL), established 60 μg/m2/d as the MTD after assessing seven different doses ranging from 0.5 to 90 μg/m2/day for 4 or 8 consecutive weeks [48]. Final results presented in 2013 included an overall response rate (RR) of 69 % (highest among follicular lymphomas) and a complete remission (CR) rate of 37 % among the 35 patients who received treatment at this MTD [48]. The median duration of response was 404 days. The dose-finding portion of a phase I/II trial of 41 pediatric and adolescent B-ALL patients (primary refractory, n = 9; relapse after alloSCT, n = 25) using blinatumomab, given as a CIVI over 4 out of every 6 weeks, identified the MTD as 15 μg/m2/day, but the phase II recommended dose was a stepwise dose of 5–15 μg/m2/day to reduce the risk of CRS [49].
Phase II trials
In the phase II portion of the previously mentioned phase I/II trial involving blinatumomab administered as a stepwise dose of 5–15 μg/m2/day to children and adolescents with B-ALL, 15 (37 %) patients acheived CR, 12 of whom were MRD-negative, and there were 3 (7 %) who had partial remissions (PR) within the first two cycles [49]. Eight (53 %) of the patients who attained CR were able to proceed to alloSCT. The German Multicenter Study Group for Adult Acute Lymphoblastic Leukemia (GMALL) group assessed blinatumomab in adult B-ALL patients (Ph-pos or neg) with MRD-positivity in a multicenter, single-arm, phase II trial (n = 21) (NCT00560794) [50]. Based on the results of earlier studies, blinatumomab was administered at 15 μg/m2/day as a CIVI over 4 to 8 weeks. Sixteen of the 20 (80 %) evaluable patients in the trial became MRD negative after 1 cycle of treatment (including 3 out of 5 Ph-pos patients), and 12 of these MRD responders had been molecularly refractory to previous therapy. The estimate for relapse-free survival (RFS) probability was 78 % at a median follow-up of 405 days [50]. After a median follow-up of 33 months, the hematologic-free survival of the 20 evaluable patients was 61 and 65 % among 9 patients who underwent alloSCT after receiving blinatumomab [51]. Four of the 6 Ph-neg MRD responders remained in hematologic and molecular remission at the time of the report.
Among adult patients with R/R Ph-neg pre-B-ALL, a German multicenter, exploratory, single-arm, phase II trial (n = 36) (NCT01209286) was performed as a dose-finding stage followed by a dose-extension stage [52]. Blinatumomab was administered by CIVI over 4 out of every 6 weeks in the hospital for the first week of the first cycle and subsequently for cycle 1 and further cycles as an outpatient. Patients had primary refractory disease or had experienced relapse after induction, consolidation, or alloSCT. Patients with a history of active CNS disease were excluded. All patients in the study had received prior standard induction therapy, and 42 % had relapsed after alloSCT. The median age was 32 years, and two patients did in fact have Ph-pos disease. The dose-finding stage assessed three sequential dose cohorts (n = 18) and determined the stepwise dose of 5–15 μg/m2/day to be the optimal dose for the dose-extension stage, in which an additional 18 patients were treated. 25 patients (69 %) achieved CR or CR with partial recovery of peripheral blood counts (CRh), 88 % of whom attained MRD-negativity. Those with more heavily pretreated disease and who had undergone alloSCT had a lower rate of CR/CRh than those patients who were in treated in first salvage. Median OS and RFS were 9.8 months (median follow-up 12.1 months) and 7.6 months (median follow-up 9.7 months), respectively. The median OS was statistically significantly higher among patients who had not relapsed after prior alloSCT (14.1 months) versus those had had undergone prior alloSCT (8.8 months). Thirteen of the responders (52 %) underwent alloSCT after achieving CR/CRh, but 6 died of treatment-related mortality, and 2 patients relapsed. Three patients in the study relapsed with CD19-negative disease [52].
Topp et al. published results of a large (n = 189), multinational, confirmatory, single-arm, phase II study (NCT01466179) that included R/R adult B-ALL patients with a median age of patients was 38 years, and approximately one-third of the patients had undergone alloSCT [53]. Eligibility criteria similar to those in the exploratory trial were applied, and blinatumomab was administered by CIVI over 4 out of every 6 weeks at 9 μg/day for the first 7 days (of cycle 1 only) and 28 μg/day thereafter. Eighty one (43 %) of the 189 patients attained CR (n = 63) or CRh within 2 cycles of therapy. Sixty four (79 %) of these responders achieved CR/CRh during cycle 1. The CR/CRh rate was 73 % for those patients with less than 50 % marrow involvement at baseline and 29 % for those with greater than 50 % involvement. Ninety (48 %) patients experienced no response, and 18 (10 %) were not evaluable. Of the 82 patients who experienced CR/CRh, 37 (45 %) were still in remission at a median of 8.9 months, and 32 (40 %) proceeded to alloSCT. The 100-day mortality post-alloSCT was 11 %. The median OS among all 189 was 6.1 months, but when accounting for MRD-negativity, which was noted in 60 (82 %) of 73 evaluable patients, OS was 11.5 months versus 6.7 months for MRD-positive patients [53].
Given the poorer outcomes observed among patients in CR with MRD-positivity versus MRD-negativity, and considering that CD19 is present on very early stages of B-cell development, there is strong rationale to assess blinatumomab’s role in treating MRD-positive disease, even among patients in hematologic CR. Results from the BLAST study, a single arm, multinational phase II trial (NCT01207388) that assessed blinatumomab at 15 μg/m2/day as a CIVI over 4 weeks every 6 weeks in R/R MRD-positive B-ALL adult patients (n = 116), were presented in December 2014 [54]. The median age of patients was 45 years, and 65 % of the patients were in first CR at the time of enrollment. As of February 2014, 79 patients were still alive and being followed. Seventy four had completed treatment (4 cycles or 1 cycle followed by alloSCT), and 32 had discontinued treatment for various reasons (3 patients were excluded from the efficacy analysis). The complete MRD response, the primary endpoint of the study, was 78 % (88 patients) after 1 cycle of treatment, and 2 additional patients had a complete MRD response after >1 cycle of treatment. Therefore, the complete MRD response rate overall was 80 %, and this did not significantly differ based on age, gender, line of therapy, or MRD burden category [54].
Ongoing clinical trials with blinatumomab
Currently, the previously discussed phase II trials NCT01209286, NCT01466179, and NCT01207388 are ongoing but not actively recruiting participants. Two other ongoing studies that are not actively recruiting is the Alcantara study, a phase II, single arm trial to evaluate the efficacy of blinatumomab in adults with R/R Ph-pos ALL (NCT02000427) and an open label trial assessing the drug’s efficacy for R/R DLBCL (NCT01741792). The National Cancer Institue (NCI)-sponsored phase II trial S1318 is assessing blinatumomab in elderly patients with Ph-neg or Ph-pos B-ALL with chemotherapy or dasatinib, respectively (NCT02143414). The NCI-sponsored phase III trial E1910 (NCT02003222) is randomizing newly diagnosed Ph-neg B-ALL adult patients to combination chemotherapy with blinatumomab versus induction chemotherapy alone. The TOWER study is a phase III, randomized, open label study investigating blinatumomab versus standard of care chemotherapy in adults with R/R B-ALL (NCT02013167), with OS as the primary endpoint. There is also an active NCI-sponsored, risk-stratified, randomized phase III study (NCT02101853) of blinatumomab in first relapse of childhood B-ALL.
Safety
In the MT103 1/01 phase I trials using the short-term infusion schedule, neurologic AEs that included aphasia, seizure, and disorientation led to discontinuation of the drug in 6 out the 21 patients in the 3 trials [33]. Infections, pyrexia, rigors, fatigue, and changes in hematologic and coagulation parameters were also noted in these trials [33]. The investigators from the phase I trial of patients with R/R NHL (n = 76) reported an overall incidence of grade 4 AEs of 66 and 4 % for grade 5 AEs, regardless of causality. There was a 71 % incidence of central nervous system (CNS) AEs, none of which were grade 4 or 5 [48]. In the pediatric B-ALL phase I/II trial discussed previously, the most common AEs were pyrexia, headache, hypertension, and anemia. Almost half of all patients experienced at least one CNS event such as tremor or confusional state, but all except one were grade 1 or 2 [49]. In the phase II GMALL group study (NCT00560794) that assessed blinatumomab in 21 adult B-ALL patients with MRD-positivity, the most common AEs were pyrexia, chills, hypogammaglobulinemia, and hypokalemia. Eighty one percent of patients experienced grade 3 or 4 AEs, the most common of which was lymphopenia (33.3 %) [50]. One patient had a grade 3 epileptic seizure during the first cycle, and another patient experienced syncope with convulsion.
In the exploratory phase II trial (n = 36) (NCT01209286), pyrexia (81 %), fatigue (50 %), headache (47 %), tremor (36 %), and leukopenia (19 %) were the most common AEs, although most were transient [52]. Infections constituted 33 % of serious adverse events (SAEs) and led to 6 deaths. One of these deaths was caused by a disseminated fungal infection of the brain of a patient who had undergone alloSCT prior to treatment. It was considered possibly related to blinatumomab, prompting the requirement for fungal prophylaxis for all relapsed alloSCT recipients who had a history of graft-versus-host disease. Nervous system or psychiatric disorders occurred in six patients, three of whom experienced signs of encephalopathy manifesting with tremor, aphasia, and confusion, and the other three of whom had epilepsy or convulsions. Two patients with a leukemic bone marrow burden of close to 90 % experienced grade 4 cytokine release syndrome (CRS), leading to the mandate that patients with a high leukemic burden receive pretreatment with dexamethasone and cyclophosphamide. Interestingly, these two patients attained a CR, and none of the nonresponding patients experienced CRS [52].
The most common AEs in the large (n = 189), confirmatory phase II study (NCT01466179) included pyrexia, which was managed with paracetamol or dexamethasone (or both), headache, febrile neutropenia, hypokalemia, and peripheral edema [53]. Disseminated intravascular coagulation occurred in 2 % of patients as did CRS in 2 % of patients. Treatment was discontinued among 10 % of patients due to AEs that were thought to be blinatumomab-related. Twenty three (12 %) patients had fatal AEs, most of which were infection-related, and three of these were possibly attributable to blinatumomab. Fifty two percent of all the patients in the study had neurologic events, although most (76 %) were grade 1 or 2, occurred mostly (87 %) in cycle 1, and were managed with dexamethasone treatment without treatment interruption. Grade 3 neurologic events occurred in 20 patients, most of whom recovered but 3 of whom died of unrelated causes after the neurological event. There were no fatal neurological AEs in this trial [53].
In the BLAST trial (n = 116) discussed earlier, common AEs included pyrexia (88 %), headache (38 %), tremor (29 %), chills (25 %), fatigue (24 %), nausea (22 %) and vomiting (22 %). 60 % of all patients experienced SAEs, including pyrexia (15 %), tremor (7 %), aphasia (5 %), encephalopathy (5 %) and overdose (5 %). There was also a fatal case of atypical pneumonia attributable to blinatumomab [53].
In the U.S., there is a Risk Evaluation and Mitigation Strategy (REMS) in place for blinatumomab, and U.S. prescribing information has a black box warning regarding the potential for neurotoxicity and CRS [44]. In an effort to mitigate some of the serious toxicities, the package insert recommends step-wise dosing of 9 μg/day on Days 1–7 and 28 μg/day on Days 8–28 of the first cycle for patients at least 45 kg in weight. Hospitalization for the first 9 days of the first cycle and for the first 2 days of the second cycle is also recommended, in addition to premedicating with dexamethasone 20 mg intravenously 1 h prior to the first dose of each cycle, prior to a step dose (such as day 8 of cycle 1), or when restarting an infusion after an interruption of 4 or more hours [44].
Indication
Blinatumomab was Food and Drug Administration (FDA)-approved under accelerated approval for the treatment of R/R, Ph-neg, precursor B-ALL in December 2014. Continued approval for this indication may depend upon confirmation of clinical benefit in subsequent trials.
Discussion and conclusions
Blinatumomab represents an important addition to the burgeoning armamentarium of therapies being developed to treat B-cell malignancies. Its impressive molecular response rates in heavily pretreated patients justify its recent FDA approval for the treatment of R/R Ph-neg ALL in adults. This agent’s activity also highlights the clinical effectiveness of bispecific antibodies that harness the power of one’s own T-cells, via a mechanism that differs from that of immune checkpoint inhibitors and chimeric antigen receptor T-cells [55].
There are many limitations to blinatumomab, however. The median duration of response and OS data obtained thus far, although impressive compared to other agents in such a heavily pretreated population, are still dismal by most people’s standards. Some have suggested that leukemia initiating cells may lack expression of CD19, thereby conferring resistance of these cells to blinatumomab and allowing for recurrence even after seemingly deep responses have been attained by this drug [56]. Therefore, its optimal role in the treatment of ALL may be in combination with chemotherapy or other novel agents, concurrently or sequentially. A trial involving the frontline treatment of elderly patients with blinatumomab in combination with chemotherapy is currently enrolling (NCT02143414).
Despite that CD19 expression is necessary for benefit of this drug, as noted in preclinical studies [38], a predictive biomarker among CD19+ disease to explain primary refractoriness to this therapy has not yet been identified. For disease progression that occurs after an initial response to therapy, CD19-negative escape, as manifested by CD19-negative clones and down-regulation of CD19-expression, may be a contributing factor [23]. This phenomenon has been observed in other CD19-directed therapies, and data are lacking to explain this mechanism of relapse [23]. The practical limitations of blinatumomab’s short half-life, necessitating long-term CIVI, its cost (as with all newly approved oncology drugs), and the need to hospitalize patients for the first and second cycles also pose some realistic challenges. It is also not known whether the drug will significantly penetrate the blood brain barrier to reach the CNS, a common sanctuary site for ALL.
Nonetheless, as already discussed, blinatumomab serves as proof of principle of the ability to exploit one’s immune system for targeting specific cell surface antigens of human malignancies. We are eager to witness the further development of this drug for the treatment of ALL and other B-cell malignancies.
References
Dores M, Devesa SS, Curtis RE, Linet MS, Morton LM (2012) Acute leukemia incidence and patient survival among children and adults in the United States. Blood 119(1):2001–7
Siegel R, Ma J, Zou Z, Jemal A (2014) Cancer statistics, 2014. CA Cancer J Clin 64(1):9–29
Vardiman JW, Arber DA, Brunning RD et al (2009) The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia : rationale and important changes. Blood 114(5):937–51
Moorman AV, Harrison CJ, Buck GA et al (2007) Karyotype is an independent prognostic factor in adult acute lymphoblastic leukemia (ALL): analysis of cytogenetic data from patients treated on the Medical Research Council (MRC) UKALLXII/Eastern Cooperative Oncology Group (ECOG) 2993 trial. Blood 109(8):3189–97
NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) Acute Lymphoblastic Leukemia [Internet]. NCCN 2014. Available from: http://www.nccn.org/professionals/physician_gls/pdf/all.pdf
Thomas DA, O’Brien S, Faderl S et al (2010) Chemoimmunotherapy with a modified hyper-CVAD and rituximab regimen improves outcome in de novo Philadelphia chromosome-negative precursor B-lineage acute lymphoblastic leukemia. J Clin Oncol 28(24):3880–9
Hoelzer D, Huettmann A, Kaul F et al (2010) Immunochemotherapy with rituximab improves molecular CR rate and outcome in CD20+ B-lineage standard and high risk patients; results of 263 CD20+ patients studied prospectively in GMALL study 07/2003. ASH Annu Meet Abstr 116(21):170
Ribera J-M (2013) Optimal approach to treatment of patients with Philadelphia chromosome-positive acute lymphoblastic leukemia: how to best use all the available tools. Leuk Lymphoma 54(1):21–7
Pulte D, Redaniel MT, Jansen L, Brenner H, Jeffreys M (2013) Recent trends in survival of adult patients with acute leukemia: overall improvements, but persistent and partly increasing disparity in survival of patients from minority groups. Haematologica 98(2):222–9
Pui C-H, Evans WE (2013) A 50-year journey to cure childhood acute lymphoblastic leukemia. Semin Hematol 50(3):185–96, Elsevier
Stock W, La M, Sanford B et al (2008) What determines the outcomes for adolescents and young adults with acute lymphoblastic leukemia treated on cooperative group protocols? A comparison of Children’s Cancer Group and Cancer and Leukemia Group B studies. Blood 112(5):1646–54
Ten Cate B, de Bruyn M, Wei Y, Bremer E, Helfrich W (2010) Targeted elimination of leukemia stem cells; a new therapeutic approach in hemato-oncology. Curr Drug Targets 11(1):95–110
Bruggemann M, Raff T, Kneba M (2012) Has MRD monitoring superseded other prognostic factors in adult ALL? Blood 120(23):4470–81
Fielding AK, Richards SM, Chopra R et al (2007) Outcome of 609 adults after relapse of acute lymphoblastic leukemia (ALL); an MRC UKALL12/ECOG 2993 study. Blood 109(3):944–50
O’Brien S, Thomas D, Ravandi F et al (2008) Outcome of adults with acute lymphocytic leukemia after second salvage therapy. Cancer 113(11):3186–91
Dombret H, Cluzeau T, Huguet F, Boissel N (2014) Pediatric-like therapy for adults with ALL. Curr Hematol Malig Rep. doi:10.1007/s11899-014-0210-9
Wetzler M, Thomas DA, Wang ES et al (2013) Phase I/II trial of nanomolecular liposomal annamycin in adult patients with relapsed/refractory acute lymphoblastic leukemia. Clin Lymphoma Myeloma Leuk 13(4):430–4, Elsevier Inc
Robak P, Robak T (2013) Older and new purine nucleoside analogs for patients with acute leukemias. Cancer Treat Rev 39(8):851–61, Elsevier Ltd
Ai J, Advani A (2014) Current status of antibody therapy in ALL. Br J Haematol. doi:10.1111/bjh.13205, 1–10
Advani AS (2013) New immune strategies for the treatment of acute lymphoblastic leukemia: antibodies and chimeric antigen receptors. Hematol Am Soc Hematol Educ Program 2013:131–7
Chevallier P, Huguet F, Raffoux E et al (2014) Vincristine, dexamethasone and epratuzumab for older relapsed/refractory CD22+ B-acute lymphoblastic leukemia patients: a Phase 2 Study. Haematologica 100(4):e128–31
Grupp SA, Kalos M, Barrett D et al (2013) Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med 368(16):1509–18
Maude SL, Frey N, Shaw PA et al (2014) Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 371(16):1507–17
Depoil D, Fleire S, Treanor BL et al (2008) CD19 is essential for B cell activation by promoting B cell receptor-antigen microcluster formation in response to membrane-bound ligand. Nat Immunol 9(1):63–72
Nadler LM, Anderson KC, Marti G et al (1983) B4, a human B lymphocyte-associated antigen expressed on normal, mitogen-activated, and malignant B lymphocytes. J Immunol 131:244–50
Wang K, Wei G, Liu D (2012) CD19: a biomarker for B cell development, lymphoma diagnosis and therapy. Exp Hematol Oncol 1(1):36
Carter RH, Fearon DT (1992) CD19: lowering the threshold for antigen receptor stimulation of B lymphocytes. Science 256:105–7
Van Zelm MC, Reisli I, van der Burg M et al (2006) An antibody-deficiency syndrome due to mutations in the CD19 gene. N Engl J Med 354:1901–12
Raponi S, De Propris MS, Intoppa S et al (2011) Flow cytometric study of potential target antigens (CD19, CD20, CD22, CD33) for antibody-based immunotherapy in acute lymphoblastic leukemia: analysis of 552 cases. Leuk Lymphoma 52(June):1098–107
Uckun FM, Jaszcz W, Ambrus JL et al (1988) Detailed studies on expression and function of CD19 surface determinant by using B43 monoclonal antibody and the clinical potential of anti-CD19 immunotoxins. Blood 71(1):13–29
Uckun FM, Manivel C, Arthur D et al (1992) In vivo efficacy of B43 (anti-CD19)-pokeweed antiviral protein immunotoxin against human pre-B cell acute lymphoblastic leukemia in mice with severe combined immunodeficiency. Blood 79:2201–14
Mitchell P, Lee F-T, Hall C et al (2003) Targeting primary human Ph(+) B-cell precursor leukemia-engrafted SCID mice using radiolabeled anti-CD19 monoclonal antibodies. J Nucl Med 44:1105–12
Nagorsen D, Kufer P, Baeuerle PA, Bargou R (2012) Blinatumomab: a historical perspective. Pharmacol Ther 136(3):334–42, Elsevier Inc
Zimmerman Z, Maniar T, Nagorsen D (2014) Unleashing the clinical power of T cells: CD19/CD3 bi-specific T cell engager (BiTE®) antibody construct blinatumomab as a potential therapy. Int Immunol 27(1):31–7
Staerz UD, Kanagawa O, Bevan MJ (1985) Hybrid antibodies can target sites for attack by T cells. Nature 314(6012):628–31, England
Mack M, Riethmüller G, Kufer P (1995) A small bispecific antibody construct expressed as a functional single-chain molecule with high tumor cell cytotoxicity. Proc Natl Acad Sci U S A 92(July):7021–5
Frankel SR, Baeuerle PA (2013) Targeting T cells to tumor cells using bispecific antibodies. Curr Opin Chem Biol 17(3):385–92, Elsevier Ltd
Dreier T, Lorenczewski G, Brandl C et al (2002) Extremely potent, rapid and costimulation-independent cytotoxic T-cell response against lymphoma cells catalyzed by a single-chain bispecific antibody. Int J Cancer 100(6):690–7
Hoffmann P, Hofmeister R, Brischwein K et al (2005) Serial killing of tumor cells by cytotoxic T cells redirected with a CD19-/CD3-bispecific single-chain antibody construct. Int J Cancer 115(1):98–104
Rabinovich GA, Gabrilovich D, Sotomayor EM (2007) Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol 25:267–96
Schub A, Nagele V, Zugmaier G, et al (2013) Immunopharmacodynamic response to blinatumomab in patients with relapsed/refractory B-precursor acute lymphoblastic leukemia (ALL). J Clin Oncol (suppl; abstr 7020)
Brandl C, Haas C, D’Argouges S et al (2007) The effect of dexamethasone on polyclonal T cell activation and redirected target cell lysis as induced by a CD19/CD3-bispecific single-chain antibody construct. Cancer Immunol Immunother 56(10):1551–63
Dreier T, Baeuerle PA, Fichtner I et al (2003) T cell costimulus-independent and very efficacious inhibition of tumor growth in mice bearing subcutaneous or leukemic human B cell lymphoma xenografts by a CD19-/CD3- bispecific single-chain antibody construct. J Immunol 170(8):4397–402
Onyx Pharmaceuticals (2015) BLINCYTO (blinatumomab) Prescribing Information
Wu B, Hijazi Y, Wolf A, Brandl C, Sun Y-N, Zhu M (2013) Pharmacokinetics (PK) of blinatumomab and its clinical implications. ASCO Meet Abstr 31(15_suppl):3048
Nagorsen D, Baeuerle PA (2011) Immunomodulatory therapy of cancer with T cell-engaging BiTE antibody blinatumomab. Exp Cell Res 317(9):1255–60, Elsevier Inc
FDA Drug Approval Package [Internet]. [cited 2015 Mar 2]. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/125557Orig1s000TOC.cfm
Goebeler M-E, Viardot A, Kufer P, Topp MS, Knop S, Mackensen L (2013) Final results from a phase 1 study of blinatumomab in patients with relapsed/refractory Non-Hodgkin’s lymphoma. Hematol Oncol 31(Abstract 302):197
Locatelli F, Gore L, Zugmaier G, Handgretinger R, Rizzari C, Trippett T (2014) Blinatumomab in pediatric patients with Relapsed/Refractory (R/R) B-cell Precursor Acute Lymphoblastic Leukaemia (BCP-ALL): a phase I/II study. Bone Marrow Transplant 37(Abstract PH-P545):S361
Topp MS, Kufer P, Gökbuget N et al (2011) Targeted therapy with the T-cell-engaging antibody blinatumomab of chemotherapy-refractory minimal residual disease in B-lineage acute lymphoblastic leukemia patients results in high response rate and prolonged leukemia-free survival. J Clin Oncol 29(18):2493–8
Topp MS, Gökbuget N, Zugmaier G et al (2012) Long-term follow-up of hematologic relapse-free survival in a phase 2 study of blinatumomab in patients with MRD in B-lineage ALL. Blood 120(26):5185–7
Topp MS, Gokbuget N, Zugmaier G et al (2014) Phase II trial of the anti-CD19 bispecific T cell-engager blinatumomab shows hematologic and molecular remissions in patients with relapsed or refractory B-precursor acute lymphoblastic leukemia. J Clin Oncol 32(36):4134–40
Topp MS, Gökbuget N, Stein AS et al (2014) Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol 15:57–66
Goekbuget N, Dombret H, Bonifacio M et al (2014) BLAST: a confirmatory, single-Arm, phase 2 study of blinatumomab, a Bispecific T-cell Engager (BiTE®) antibody construct, in patients with minimal residual disease B-precursor Acute Lymphoblastic Leukemia (ALL). Blood 124(21):Abstract 379
Ruella M, Gill S (2015) How to train your T cell: genetically engineered chimeric antigen receptor T cells versus bispecific T-cell engagers to target CD19 in B acute lymphoblastic leukemia. Expert Opin Biol Ther 15(6):761–6
Thomas X (2014) Blinatumomab: a new era of treatment for adult ALL? Lancet Oncol 16(1):6–7, Elsevier Ltd
Disclosures
We have no conflicts of interest to disclose.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Kaplan, J.B., Grischenko, M. & Giles, F.J. Blinatumomab for the treatment of acute lymphoblastic leukemia. Invest New Drugs 33, 1271–1279 (2015). https://doi.org/10.1007/s10637-015-0289-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-015-0289-4